c-메트 억제제
c-Met inhibitorc-Met 억제제는 간세포 성장 인자/스캐터 인자(HGF/SF)의 수용체인 c-Met tyrosine kinase의 효소 활성을 억제하는 작은 분자의 일종이다. 이러한 억제제는 다양한 종류의 암 치료에 치료적 응용이 있을 수 있다.[1]
많은 c-Met 억제제들이 현재[when?] 임상시험 중이다. Crizotinib[2]과 cabozantinib는 동안 cabozantinib 2012년에 속질 갑상선 cancer[3]의 치료 목적으로 그것은 또한 임상 실험 시작했다 통과시킨 미국 FDA.Crizotinib에 의해 승인되기를 첫번째 2011년 로컬 또는 전이성 발전해 중소 세포 폐 암 환자들의 치료를 받기 위해 가속 승인을 받은 것. 카메라에그는 몇 가지 다른 종류의 암을 치료한다.
c-Met은 세포 산란, 침공, 세포사멸 및 혈관신생을 촉진시킨다.[4] c-Met은 수용체 티로신 키나아제로,[5] 신장, 위암, 소세포폐암, 중추신경계 종양 등 다양한 암을 유발할 수 있으며, 활동이 조절되지 않을 경우 여러 개의 사포를[6] 유발할 수 있다. 작은 분자 억제제에 의한 c-Met의 ATP 결합 부위를 목표로 하는 것은 티로신 키나아제의 억제를 위한 하나의 전략이다.[7]
역사

1980년대 초 MET는 변형된 종양 유전자의 단백질 산물로 설명되었다.[9] [10]


2002년 ATP 경쟁 c-Met 억제제를 식별하려는 초기 시도는 k252a의 발견으로 이어졌는데, 이는 c-Met를 차단하는 staurosporine 유사 억제제로서 K252a는 비인산 MET kinase 도메인과 복잡하게 해결된 첫 번째 구조였다.[10][11] 그것은 힌지와 피랄로카르바졸 서브 유닛 사이에 두 개의 수소 결합을 형성한다.[8]
이후에 일련의 선택적 c-Met 억제제가 설계되었으며, 여기서 인돌린-2-1 코어(그림 1에 동그라미)가 여러 키나제 억제제에 존재하였다. SU-11274는 인돌리논의 5개 위치에서 대체하여 진화하고 3,5-디메틸 피롤 그룹을 추가하여 PHA-665752를 진화시켰으며, 효력과 활성도가 더 높은 2세대 억제제였다.[10]
이 분야에 대한 관심은 2007년 이후 급격히 높아져 2009년 중반에 70여 건의 특허 출원이 발표되었다.[10]
c-Met가 암치료의 적정한 대상으로 받아들여진 데 이어 제약업계에서도 집중적인 노력이 기울여지고 있다. 리간드가 있거나 없는 20개의 결정 구조가 출판되었고 2010년에 거의 12개의 작은 분자 c-Met 억제제가 임상적으로 시험되었다.[12]
소개
수용체 Tyrosine kinases(RTKs)는 많은 세포내 신호 전달 경로를 조절하는 데 필수적인 요소다.[13] Met tyrosine kinase는 간세포 성장인자(HGF)의 수용체로, 산란인자(SF)로도 알려져 있다. HGF는 주로 상피세포와 중피세포(예: 부드러운 근육세포와 섬유소)에 표현된다.[10][11] HGF는 보통 상처 치유, 간 재생, 배아 및 정상적인 포유류 발달,[10] 장기 형태생식에 적극적이다.[11]
c-메트 조절장애는 과도한 압박, 유전자 증폭, 돌연변이, 리간드에 의존하는 자동 또는 파라신 루프 또는 RTK의 시기적절한 활성화에 기인할 수 있다.[10][13] 이 모든 요소들은 세포의 생존, 그 증식과 운동성에 영향을 미친다. 그들은 또한 암과 그들을 치료하는 것을 목표로 하는 치료법에 대한 내성을 초래한다.[13] 이상 c-Met 활동을 하는 환자들은 대개 예후가 좋지 않고, 공격적인 질병, 전이가 증가하고, 생존이 짧아진다.[10] 이 때문에 HGF/c-MET 신호 전달 경로의 타겟팅이 암 치료로서 이루어지지 않고 있으며,[10][13] 여러 가지 다른 치료적 접근법이 임상적으로 시험되고 있는 것이다. 각각 항체, 펩타이드 작용제,[4][10] 디코이 수용체 및 기타 생물학적 억제제[14] 또는 소분자 억제제에 의한 c-Met 활성화를 규제하는 직렬 단계 중 하나에 초점을 맞춘 다양한 접근법이 c-Met를 대상으로 사용되어 왔다.[10]
구조 및 기능


c-Met RTK 하위 패밀리는 다른 많은 RTK 패밀리와 구조가 다르다. 성숙한 형태는 이황화 결합에 의해 서로 연결되는 세포외 α체인(50kDa)과 트랜섬브레인 β체인(140kDa)이 있다. 베타 체인은 세포내 타이로신 키나아제 도메인과 기판과 다운스트림 신호의 도킹에 필수적인 C-터미널의 꼬리를 포함한다.[10] [17]
HGF는 메트를 위한 자연적인 고선위 리간드다.[10][11][17] 그것의 N-단자 영역은 Met에 결합되고 수용체 조광화는 물론 두 개의 타이로인의 자기인산화도 Met의 키나아제 영역의 활성화 루프(A-루프)에서 발생한다.[10]
인산화 작용은 C-terminus에 가까운 tyrosine에서 발생하며, 어댑터 단백질을 모집하고 다운스트림 신호 전달을 유도하는 다기능 도킹 사이트를[10][18] 만든다. 이 신호는 Ras/Mapk, PI3K/Akt, c-Src 및 STAT3/5에 의해 매개되며 세포 증식, 세포골격계 기능 변경 등을 포함한다.
키나제 영역은 보통 로브가 매우 보존된 ATP 결합 부지에 인접하여 힌지 영역과 연결되는 양로 구조로 구성된다.[10]
개발
PHA-66752와 c-Met의 공결정 구조로부터의 정보를 이용하여 선택적 억제제 PF-2341066을 설계하였다. 2010년에는 임상 1상 시험을 실시하고 있었다. 4-페녹시퀴놀린 화합물을 아킬 티우레아 그룹과 바꾸면 c-Met 활성(예: 퀴놀린)과 화합물이 된다.[10] 이것은 아틸 결합이 터미널 아릴 그룹에게 깊은 소수성 포켓을 관통할 수 있는 능력을 부여하여 화합물의 효력을 향상시킨다는 점에서 c-Met 억제제 개발 진행의 핵심 단계였다. AM7에서와 같이 피리미돈 그룹을 가진 아킬 티우레아 연결에 대한 대안이 발견되었다.[19]
AM7과 SU11274는 비교적 선택적인 c-Met 억제제가 식별될 수 있고 그 억제가 체내 투약 방지 효과를 유발한다는 첫 번째 증거를 제공했다. AM7과 SU11274의 공결정 구조를 c-Met과 비교했을 때, U자형 순응으로 힌지 영역에 인접한 SU-11274 바인딩, 그러나 AM7은 힌지 영역에서 C-헬릭스까지의 영역을 포괄하는 확장 순응으로 c-Met에 바인딩하는 등 서로 다른 것으로 밝혀졌다. 그런 다음 그것은 소수성 주머니에서 결합된다. c-Met은 AM7과 함께 비활성화되고 비인산성 순응을 가정한다. AM7은 키나제의 인산염 순응과 비인산 순응 둘 다에 결합할 수 있다.[20]
이 두 가지 다른 유형의 결합 때문에, 작은 분자 Met 억제제는 클래스 I (SU-11274 유사)과 클래스 II (AM7 유사)의 두 종류로 나뉘었다.[20] 그러나 두 종류 중 어느 한 종류에도 들어맞지 않는 또 다른 유형의 소분자 억제제가 있는데, 그것은 다른 두 종류와 다른 방식으로 결합되는 비경쟁 ATP 억제제다.[21]
작은 분자 억제제는 선택성이 다양하며, 매우 구체적이거나 선택성이 넓다. 그들은 ATP 경쟁적이거나 비경쟁적이다.[12]
ATP 경쟁 소형 분자 c-Met 억제제
두 등급은 구조적으로 다르지만 다음과 같은 특성을 공유한다. 둘 다 키나세 힌지 영역에 바인딩되며(C-Met 활성 사이트의[20] 서로 다른 부분을 점유하고 있지만), 모두 ATP의 퓨린 흉내를 목표로 한다. BMS-777607과 PF-02341066은 2-아미노 피리딘 그룹을, AMG-458은 퀴놀린 그룹을, MK-2461은 트리시클릭 그룹을 가진다.[22]
1급
클래스 I 억제제는 많은 다른 구조를 가지며,[12] 상대적으로 선택적이며 U자형 순응성을[10] 가지며, c-Met의 활성화 루프에 결합된다.[12]
I급 억제제의 구조활동 관계

c-MET 억제제로서 큰 가능성을 보인 트라이아졸로트리아진(triazolotriazine)이 잇따라 발견됐다. 구조 활동 관계(SAR)는 3가지 링에 연결된 아릴 그룹과 펜던트 벤질 링에 부착된 적절한 수소 결합 수용체(예: 히드록실 그룹)의 필요성을 내포하지만 페놀은 힌지 바인더(메트1160)로 작용하고 트라이아진이 Tyr1230과 상호 작용하는 것처럼 보인다.[12] 유사한 유사점이 다수 발견되어 분석되었다. 페놀 힌지 결합 요소가 아릴라미노-트리아졸로피리다진 또는 아릴-트리아졸로티오피라빈에 연결된 구조적으로 유사한 일련의 c-Met 억제제. 1원자 링커는 2원자 링커보다 더 효율적이었고 벤자민 위치에서 대체하는 것은 용인되는 것 같았다. 이질소성 힌지 결합 요소(퀴놀린, 피리딘, 아자인돌)가 융합된 질소성 이질소성 이질소성(triazolopyridazine, triazolophyrazine, triazolotriazine)과 연결된 화합물이 설명되었다.[12] 자세한 내용은 그림 4를 참조하십시오.[12]
I급 억제제의 예
디플루오로 메틸링커와 생체이용 가능한 퀴놀린 그룹이 포함된 JNJ-38877605는 2010년 고도 및 내화성 고체 종양에 대해 1단계 임상시험을 진행 중이었다.[12] [needs update]
PF-04217903은 ATP와 경쟁하고 예외적으로 선택성이 높은 화합물로서 Triazolopyrazine의 C-7에 연결된 N-hydroxyethyl pyrazole 그룹을 가지고 있다. 그것은 2010년에 임상 1상을 거치고 있었다.[12] [needs update]
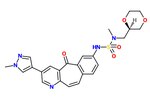
강력한 c-Met 억제 활성을 가진 고유 키나제 억제제 비계 MK-2461의 SAR를 탐구하였다.[23] 피리딘 질소는 억제 활동과 중앙 링 포화도를 감소시키는 효력 감소에 필요하다.[12] 분자의 평면성은 최대 효력에 필수적인 것으로 입증되었다.[23] 주기적인 에테르들은 수용 가능한 세포 기반 활동과 약동학적 특성 사이의 균형을 맞춘다. 최적화 프로세스에서는 다음과 같은 요소들이 핵심 요소는 다음과 같다.
1) 소수성 패킹과 평면성을 최대화하듯 7위치에 있는 아릴 그룹,
2) Sulfonamide 그룹 첨가 시 타이트한 SAR과
3) 용제노출군의 비교적 평탄한 SAR.
종종 c-Met의 종양성 돌연변이는 작은 분자 억제제에 대한 저항을 유발한다. 따라서 MK-2461 아날로그는 다양한 c-Met 돌연변이에 대해 테스트되었지만 그것들에 대해 덜 강력한 것으로 입증되었다. 이것은 분자에게 c-Met 조절 오류로 인한 종양 치료로서 큰 이점을 준다.[23] MK-2461은 2010년에 1단계 선량 에스컬레이션 시험을 거치고 있었다.[12] [needs update]
클래스 II
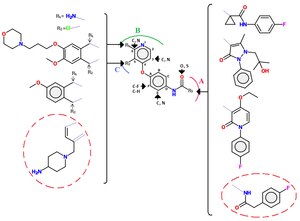
등급2 억제제는 대개 등급1 억제제만큼 선택적이지 않다.[10] 요소군은 또한 순환형 또는 순환형 형태로 분류되는 II급 억제제의 공통적인 특징이다. 억제제의 등급 II에는 그림 4에서 볼 수 있는 여러 가지 다른 분자가 포함되어 있다.[12]
II등급 억제제의 구조활동 관계
아킬티우레아 연계가 있는 일련의 퀴놀린 c-Met 억제제를 연구했다. 대체 힌지 결합 그룹(예: 퀴놀린 그룹의 교체), 티우레아 연결부(예: 말로나미드, 옥살라미드, 피라졸론)의 교체 및 다양한 방향성 헤테로사이클릭 아킬티우레아 구조 파편을 구속하는 일련의 아날로그가 발견되었다. 추가적인 미세화에는 플루오린 원자로 펜던트 페닐 링의 p 위치를 차단하는 것이 포함되었다.[12] 등급 II의 c-Met과 작은 분자(빨간색 원으로 표시됨) 사이의 상호작용의 예는 다음과 같다. c-Met의 비계는 세 가지 주요 수소 결합에 의해 ATP 포켓에 박혀 있으며, 터미널 아민은 리보스 포켓(ATP의)과 상호 작용하며, 터미널 4 플로로페닐 그룹은 소수성 포켓을 지향하며, 피롤로트리아진은 힌지 바인딩 그룹의 역할을 한다.[12]
클래스 II 억제제의 예
임상 2상에서는 GSK 1363089(XL880, poretinib)가 잘 용인되었다. 유두 신장암 환자에게는 약간의 퇴행이나 안정적 질환이 발생했고, 위암도 잘 구분되지 않았다.[12]
AMG 458은 55개의 키나아스로 구성된 패널에서 100배 이상의 c-MET 선택성을 갖는 것으로 입증된 강력한 소형 분자 c-MET 억제제다. 또한, AMG 458은 종에 걸쳐 100% 생물학적으로 이용 가능했고, 더 높은 포유동물과 함께 본질적인 반감기가 증가했다.[12]
ATP 비경쟁적 소분자 c-Met 억제제
티반티닙

이 기사는 티반티닙이라는 글과 모순되는 것으로 보인다. (2015년 11월)(이과 시기 |
티반티닙(ARQ197)은 선택적이고[when?] 경구적으로 이용할 수 있으며 [17][21]임상적으로 발달한 저분자중량 및 잘 절연된 c-MET 억제제로, 현재 비소세포 폐암 환자에게 임상 3상 임상시험 중이다.[21] ARQ197은 ATP 경쟁사 c-MET 자동인산 억제제로 키나제의 비인산성 순응에 대한 선택성이 높다.[17][21] Tivantinib은 주요 촉매 잔류물 사이의 상호작용을 차단한다.[21] c-Met kinase 도메인과 복잡한 tivantinib의 구조는 억제제가 발표된 kinase 구조와 구별되는 준수를 결합한다는 것을 보여준다. 티반티닙은 N-로브와 C-로브 사이의 키나아제의 비활성 형태를 선택적으로 대상으로 하여 c-Met 자동 활성화를 강력하게 억제하고 ATP 바인딩 사이트를 점유한다.[21]
임상시험 및 규제승인
2010년 기준 현황
메트와 HGF의 발견 이후, 많은 연구 관심은 암에서의 그들의 역할에 집중되었다. Met route는 인간 암에서 가장 빈번하게 조절이 잘 되지 않는 경로 중 하나이다.[17] 바인딩 모드와 구조 설계에 대한 이해도를 높이면 다른 단백질 상호작용과 바인딩 포켓의 사용에 더 가까워져 대체 구조와 최적화된 프로파일로 억제제를 만들 수 있다.[10]

2010년[update] 현재 12개 이상의 메트 경로 억제제, 고선택적에서 다중 표적까지의 다양한 키나아제 선택성 프로파일이 진료실에서 연구되었으며 양호한 진척이 달성되었다([12]예: XL184(Cabozantinib), XL880, ARQ197 참조).
다른 치료제들과 함께 c-Met 억제제를 사용하는 것은 잠재적 저항성을 극복하는 것은 물론 전반적인 임상 유익성 향상에 결정적일 수 있다. 메트 경로 억제제는 화학요법, 무선요법 또는 면역요법뿐만 아니라 HGF와 메트 생물학적 길항억제제 또는 HGF와 메트 생물학적 길항제나 항체를 HGF와 MET에 대한 다른 치료제와 함께 사용될 수 있다.[17] 그러나 누적 독성 및 다른 약물과 상호작용의 위험은 여전하다.[10]
2010년 이후
2011년 PF-02341066(현재 crizotinib)은 일부 비소세포 폐암에 대해 미국 FDA의 승인을 받았다.
2012년 XL184/카보잔티닙은 중상 갑상선암 치료를 위한 FDA 승인을 얻었고, 2016년에는 신장암 치료를 위한 FDA와 EU 승인을 얻었다.
기타억제제 연구
Tepotinib, (MSC 2156119J),[24]
폐암에 대한 임상 2상 결과가 보고되었다.[25] 테포티닙은 2019년 9월 미국 식품의약국(FDA)으로부터 획기적인 치료제 지정을 받았다.[26] 2019년 11월에는 일본에서, 2020년 9월에는 호주에서 고아 마약 지정을 받았다.[27]
참고 항목
외부 링크
참조
- ^ Liu X, Newton RC, Scherle PA (September 2011). "Development of c-MET pathway inhibitors". Expert Opin Investig Drugs. 20 (9): 1225–41. doi:10.1517/13543784.2011.600687. PMID 21740293. S2CID 24415851.
- ^ Kazandjian, D; et al. (Oct 2014). "FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements". Oncologist. 19 (10): e5-11. doi:10.1634/theoncologist.2014-0241. PMC 4201002. PMID 25170012.
- ^ "FDA approves Cometriq to treat rare type of thyroid cancer". 29 November 2012.
- ^ a b Comoglio PM, Giordano S, Trusolino L (June 2008). "Drug development of MET inhibitors: targeting oncogene addiction and expedience". Nature Reviews Drug Discovery. 7 (6): 504–16. doi:10.1038/nrd2530. PMID 18511928. S2CID 24601127.
- ^ Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R (February 2002). "Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition". Cytokine Growth Factor Rev. 13 (1): 41–59. doi:10.1016/S1359-6101(01)00029-6. PMID 11750879.
- ^ Davis IJ, McFadden AW, Zhang Y, Coxon A, Burgess TL, Wagner AJ, Fisher DE (January 2010). "Identification of the receptor tyrosine kinase c-Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma". Cancer Res. 70 (2): 639–45. doi:10.1158/0008-5472.CAN-09-1121. PMC 2807989. PMID 20068147.
- ^ Porter J, Lumb S, Franklin RJ, Gascon-Simorte JM, Calmiano M, Riche KL, Lallemand B, Keyaerts J, Edwards H, Maloney A, Delgado J, King L, Foley A, Lecomte F, Reuberson J, Meier C, Batchelor M (May 2009). "Discovery of 4-azaindoles as novel inhibitors of c-Met kinase". Bioorg. Med. Chem. Lett. 19 (10): 2780–4. doi:10.1016/j.bmcl.2009.03.110. PMID 19369077.
- ^ a b Schiering N, Knapp S, Marconi M, Flocco MM, Cui J, Perego R, Rusconi L, Cristiani C (October 2003), "Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a", Proc. Natl. Acad. Sci. U.S.A., 100 (22): 12654–12659, Bibcode:2003PNAS..10012654S, doi:10.1073/pnas.1734128100, PMC 240673, PMID 14559966
- ^ a b Sattler M, Pride YB, Ma P, Gramlich JL, Chu SC, Quinnan LA, Shirazian S, Liang CX, Podar K, Christensen JG, Salgia R (September 2003), "A novel small molecule Met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase", Cancer Research, 63 (17): 5462–5469, PMID 14500382
- ^ a b c d e f g h i j k l m n o p q r s t u Porter, J (February 2010), "Small molecule c-Met kinase inhibitors: a review of recent patents", Expert Opinion on Therapeutic Patents, 20 (2): 159–177, doi:10.1517/13543770903514137, PMID 20100000, S2CID 22743228
- ^ a b c d e Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, Chen J, Wang XY, Ruslim L, Blake R, Lipson KE, Ramphal J, Do S, Cui JR, Cherrington JM, Mendel DB (November 2003), "A selective small molecule inhibitor of c-Met kinase inhibits c-Met dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo", Cancer Research, 63 (21): 7345–55, PMID 14612533
- ^ a b c d e f g h i j k l m n o p q r s t u Underiner TL, Herbertz T, Miknyoczki SJ (January 2010), "Discovery of Small Molecule c-Met Inhibitors: Evolution and Profiles of Clinical Candidates", Anti-Cancer Agents in Medicinal Chemistry, 10 (1): 7–27, doi:10.2174/1871520611009010007, PMID 20015007
- ^ a b c d Sattler M, Salgia R (April 2009), "The Met axis as a therapeutic target", Update on Cancer Therapeutics, 3 (3): 109–118, doi:10.1016/j.uct.2009.01.001, PMC 2847295, PMID 20368753
- ^ Christensen JG; Burrows J; Salgia R. (July 2005), "c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention", Cancer Letters, 225 (1): 1–26, doi:10.1016/j.canlet.2004.09.044, PMID 15922853
- ^ Knudsen BS, Woude GV (February 2008), "Showering c-MET-dependent cancers with drugs", Current Opinion in Genetics & Development, 18 (1): 87–96, doi:10.1016/j.gde.2008.02.001, PMID 18406132
- ^ Donald P. Bottaro; Megan Peach; Marec Nicklaus; Terrence Burke, JR.; Gagani Athauda; Sarah Choyke; Alessio Guibellino; Nelly Tan; Zhen-Dan Shi (August 2011), "Compositions and methods for inhibition of hepatocyte growth factor receptor c-Met signaling", United States Patent Application Publication
- ^ a b c d e f g Liu XD, Newton RC, Scherle PA (January 2010), "Developing c-MET pathway inhibitors for cancer therapy: progress and challenges", Trends in Molecular Medicine, 16 (1): 37–45, doi:10.1016/j.molmed.2009.11.005, PMID 20031486
- ^ Kung PP, Funk L, Meng J, Alton G, Padrique E, Mroczkowski B (June 2008), "Structure activity relationships of quinoline-containing c-Met inhibitors", European Journal of Medicinal Chemistry, 43 (8): 1321–1329, doi:10.1016/j.ejmech.2007.08.011, PMID 17964000
- ^ Bellon SF; Kaplan-Lefko P; Yang YJ; Zhang YH; Moriguchi J; Rex K; Johnson CW; Rose PE; Long AM; O‘Connor AB; Gu Y; Coxon A; Kim TS; Tasker A; Burgess TL; Dussault I (February 2008), "c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations", Journal of Biological Chemistry, 283 (5): 2675–2683, doi:10.1074/jbc.M705774200, PMID 18055465
- ^ a b c Dussault I, Bellon SF (February 2009), "From concept to reality: the long road to c-Met and RON receptor tyrosine kinase inhibitors for the treatment of cancer", Anti-Cancer Agents in Medicinal Chemistry, 9 (2): 221–229, doi:10.2174/187152009787313792, PMID 19199866
- ^ a b c d e f Eathiraj S, Palma R, Volckova E, Hirschi M, France DS, Ashwell MA, Chan TC (June 2011), "Discovery of a Novel Mode of Protein Kinase Inhibition Characterized by the Mechanism of Inhibition of Human Mesenchymal-epithelial Transition Factor (c-Met) Protein Autophosphorylation by ARQ 197", Journal of Biological Chemistry, 286 (23): 20666–20676, doi:10.1074/jbc.M110.213801, PMC 3121448, PMID 21454604
- ^ Allen JV, Bardelle C, Blades K, Buttar D, Chapman L, Colclough N, Dossetter AG, Garner AP, Girdwood A, Lambert C, Leash AG, Law B, Major J, Plant H, Slater AM (September 2011), "The discovery of benzanilides as c-Met receptor tyrosine kinase inhibitors by a directed screening approach", Bioorganic & Medicinal Chemistry Letters, 21 (18): 5224–5229, doi:10.1016/j.bmcl.2011.07.047, PMID 21835616
- ^ a b c Katz JD, Jewell JP, Guerin DJ, Lim J, Dinsmore CJ, Deshmukh SV, Pan BS, Marshall CG, Lu W, Altman MD, Dahlberg WK, Davis L, Falcone D, Gabarda AE, Hang GZ, Hatch H, Holmes R, Kunii K, Lumb KJ, Lutterbach B, Mathvink R, Nazef N, Patel SB, Qu XL, Reilly JF, Rickert KW, Rosenstein C, Soisson SM, Spencer KB, Szewczak AA, Walker D, Wang WX, Young J, Zeng QW (June 2011), "Discovery of a 5H-Benzo[4,5]cyclohepta[1,2-b]pyridin-5-one (MK-2461) Inhibitor of c-Met Kinase for the Treatment of Cancer", Journal of Medicinal Chemistry, 54 (12): 4092–4108, doi:10.1021/jm200112k, PMID 21608528
- ^ 국소급 또는 전이성 비소세포폐암(NSCLC) 피험자에 게피티닙이 있는 Tepotinib(Tepotinib)
- ^ MET exon 14개의 건너뛰기 돌연변이를 가진 첨단 폐선두정종 c-Met 억제제 tepotinib의 2단계 실험. 2017
- ^ "Tepotinib Breakthrough Therapy". Merck KGaA, Darmstadt, Germany (Press release). 11 September 2019. Retrieved 8 November 2020.
- ^ "Orphan Drug Designation". Merck KGaA, Darmstadt, Germany (Press release). 20 November 2019. Retrieved 8 November 2020.
