니트릴
Nitrile니트릴(nitrile)은 -C≡N 기능군을 가지는 모든 유기 화합물이다.[1] cyano-라는 접두사는 산업 문학에서 nitrile이라는 용어와 바꾸어 사용된다. 질산은 슈퍼 접착제에 사용되는 메틸 시아노아크릴레이트, 라텍스 없는 실험실과 의료용 장갑에 사용되는 질소 함유 폴리머인 니틸 고무 등 많은 유용한 화합물에서 발견된다. 니트릴 고무는 연료와 기름에 내성이 있기 때문에 자동차와 다른 물개로도 널리 사용된다. 여러 니트리올 그룹을 포함하는 유기 화합물을 시아노카본이라고 한다.
-C≡N 집단을 포함하는 무기 화합물을 질산염이라고 하는 것이 아니라 시안화라고 부른다.[2] 질소와 시안화물은 모두 청산가리의 소금에서 추출할 수 있지만 대부분의 질화물은 독성이 거의 없다.
구조 및 기본 특성
N-C 지오메트리는 질소로 선형이며 삼각 결합 탄소의 sp 하이브리드화를 반영한다. C-N 거리는 1.16 å로 짧으며, 삼중 결합과 일치한다.[3] 질산은 극성이며, 높은 쌍극자 모멘트에서 알 수 있다. 액체로써, 그들은 종종 30년대에 상대적인 허용오차가 높다.
역사
아질산염의 동음이의 첫 번째 화합물인 포름산의 니트리올인 시안화수소는 1782년 C. W. Schele에 의해 처음으로 합성되었다.[4][5] 1811년 J. L. 게이 루삭은 매우 독성이 강하고 휘발성이 강한 순산을 준비할 수 있었다.[6] 1832년경 벤조니트릴(benzonitrile)은 벤조산의 질소인 벤조니틀(benzonitrile)을 프리드리히 뵐러(friedrich Wöler)와 저스투스 폰 리비히(justususus von libig)에 의해 준비되었지만, 합성의 수율이 미미했기 때문에 물리적 특성이나 화학적 특성도 제시되지 않았다. 1834년 테오필-줄스 펠루제는 프로피온 알코올과 하이드로시아산의 에테르임을 암시하면서 프로피온이트릴을 합성했다.[7] 벤조산 암모늄을 가열하여 1844년 헤르만 페흘링에 의한 벤조니트릴의 합성은 화학 연구에 필요한 물질의 충분한 양을 산출하는 첫 번째 방법이었다. 펠링은 자신의 결과를 이미 알려진 시안화수소의 합성 암모늄 포메이트를 가열하여 그 구조를 결정했다. 그는 새로 발견된 물질에 대해 "니트릴"이라는 이름을 만들었고, 이것은 이 화합물 그룹의 이름이 되었다.[8]
합성
산업적으로 질산염의 주요 생산 방법은 암산화 및 수산화다. 두 경로 모두 계량학적 양의 염분을 생성하지 않는다는 점에서 녹색이다.
암산화
암산화에서 탄화수소는 암모니아에서 부분적으로 산화된다. 이 변환은 아크릴로니트릴에 대해 대규모로 실행된다.[9]
- CHC3=CH2 + 3⁄2 O2 + NH3 → NCH=CH2 + 3 HO2
아크릴로니트릴 생산에서 사이드 제품은 아세토나이트릴이다. 산업 규모로는 벤조니트릴, 프탈로니트릴과 이소부티로니트리올의 여러 파생상품이 암산화술에 의해 준비된다. 이 과정은 금속 산화물에 의해 촉매되며 이미인을 통해 진행되는 것으로 가정한다.
하이드로시아네이션
하이드로시아네이션은 시안화수소와 알켄화수소로부터 질소를 생산하기 위한 산업적인 방법이다. 이 공정에는 균일한 촉매가 필요하다. 하이드로사이아닐의 생산은 1,3-부타디엔에서 나일론-6,6의 전구체인 아디포니트릴의 생산이다.
- CH2=CH-CH=CH2 + 2 HCN → NC(CH2)4씨엔
유기 할로겐화물과 시안화염으로부터
두 가지 염전반응은 실험실 체중계 반응에 인기가 있다. 콜베 니트릴 합성에서는 알킬 할로겐화물이 알칼리 금속 시안화물과 함께 핵포필성 알리프화 대체물을 거친다. 아릴 니트릴은 로젠문트-본 브라운 합성에 준비되어 있다.
키아노리딘

시아노 무수체는 특별한 종류의 질산염이다. 고전적으로 그것들은 알칼리 금속 시안화물이 시아노 무수신 반응에서 알데히드에 첨가되어 발생한다. 유기 카보닐의 극성 때문에 이 반응은 알켄의 수산화와는 달리 촉매를 필요로 하지 않는다. O-실릴 시아노 무수체는 촉매(실리실시란화)가 존재하는 곳에서 시안화 트리메틸을 첨가하여 생성된다. 시아노 무수 또한 아세톤 시아노 무수인을 HCN의 원천으로 하는 등 시아노 무수인 반응에 의해 준비된다.[10]
아미드 탈수
질산은 일차 아미드의 탈수로 인해 준비될 수 있다. 이를 위한 일반적인 시약으로는 오산화 인(PO25)[11]과 염화 티오닐(SOCl2)이 있다.[12] 관련된 탈수증에서, 2차 아미드는 폰 브라운 아미드 분해에 의해 질소를 준다. 이 경우 C-N 본드 1개가 갈라진다.

소변을 통해 알데히데스로부터
알데히드를 아산화질소로 변환하는 것이 인기 있는 실험실 경로다. 알데히드는 주변 온도만큼 낮은 온도에서 히드록시아민 염과 쉽게 반응하여 증감 시간을 준다. 트리에틸아민/황산가스, 제올라이트 또는 염화황화물을 포함한 광범위한 시약이 이를 지원할 수 있지만,[13] 이러한 시약은 간단한 가열로 질소로 건조될 수 있다. 관련 히드록시아민-O-술폰산이 유사하게 반응한다.[14]

알리프티알독시메탈 탈수효소와 같은 생체촉매도 사용할 수 있다.
샌드마이어 반응
방향성 질산은 종종 아닐린에서 디아조늄 화합물을 통해 실험실에 준비된다. 이것은 샌드마이어의 반응이다. 전이 금속 시안화물이 필요하다.[15]
- ArN+
2 + CuCN → ARCN + N + Cu2+
기타 방법
- 청산가리의 상업적 공급원은 디에틸알루미늄 시안화 에탈CN으로 트리틸알루미늄과 HCN에서 조제할 수 있다.[16] 그것은 케톤에 핵동위원소를 첨가하는 데 사용되어 왔다.[17] 그 사용의 예는 쿠와지마 택솔 총합성을 참조한다.
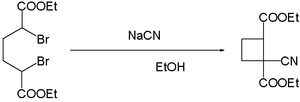
- 시안 이온은 디브로미드의 결합을 촉진한다. 에탄올에 시안화나트륨이 함유된 α,α′-디브로모아디프산의 반응은 시아노 사이클로부탄(Cyano Cylobutane)을 산출한다.[18]
- 이른바 프랜차이몬트 반응(벨기에 박사과정 학생 앙투안 폴 니콜라스 프랜차이몬트(1844-1919)가 1872년에 개발한 것)에서 α-브로모카르복실산은 시아노그룹과 디카르복실화의[19] 가수분해 후 희석된다.

- 방향족 질산은 트리클로로메틸아릴케티민(RC(CCl3)=)의 염기 가수분해로부터 조제할 수 있다.NH) Huben-Fischer 합성[20] 시
- 질산은 산화를 통해 1차 아민에서 얻을 수 있다. 일반적인 방법으로는 페르설프산 칼륨,[21][22] 트리클로로아오시아누르산 또는 양극성 전기합성이 있다.[23]
- α-아미노산은 산화 데카르복시화의 다양한 수단을 통해 질소와 이산화탄소를 형성한다.[24][25] 헨리 드라이스데일 다킨은 1916년에 이 산화를 발견했다.[26]
- 아릴 카복실산(렛츠 니트릴 합성)으로부터
반응
유기화합물 내 니트릴 그룹은 반응제나 조건에 따라 다양한 반응을 겪을 수 있다. 니트릴 그룹은 시안 이온으로서 분자를 가수 분해, 감소 또는 배출할 수 있다.
가수 분해
질소 RCN의 가수분해효과는 산 또는 염기 처리 하의 뚜렷한 단계에서 진행되어 우선 카복사미드 RC(=O)를 부여한다.NH와2 카복실산 RCOOH. 질소를 카르복실산에 가수분해하는 것은 효율적이다. 산 또는 염기에서 균형 방정식은 다음과 같다.
- RCN + 2H2O + HCl → RCO2H + NH4Cl
- RCN + HO2 + NaOH → RCONA2 + NH3
엄밀히 말하면 산이나 염기 중 하나가 암모늄 또는 카르복실산염을 형성하기 위해 소비되기 때문에 이러한 반응은 산이나 염기에 의해 매개된다는 점에 유의한다.
운동 연구는 아세토나이트릴에서 아세타미드까지의 수산화물 촉매 가수분해를 위한 2차 비율 상수가 1.6 × 10−6 M−1 초로−1, 아미드가 카복실산물에 대한 가수분해(7−5.4−1 × 10 M s−1)보다 느리다는 것을 보여준다. 따라서 염기 가수 분해 경로는 카복시산(또는 카복시산염에 오염된 아미드)을 공급할 수 있다. 반면 산성 촉매반응은 용해체의 발열 특성에 의해 촉진되는 폴리머 형성을 피하기 위해 온도와 시약 비율에 대한 세심한 통제가 필요하다.[27] 니트리올을 해당 1차 아미드로 변환하는 고전적인 절차는 니트리올을 차가운 농축 황산에 첨가하는 것을 요구한다.[28] 카복실산으로의 추가적인 전환은 낮은 온도와 낮은 농도의 물로 인해 바람직하지 않다.
- RCN + H2O → RC(O)NH2
두 종류의 효소는 질산염의 가수분해를 촉진한다. 니트릴레이즈는 질소를 카르복실산에 가수 분해한다:
- RCN + 2 HO2 → RCOH2 + NH3
니트리올 하이드라타제는 아미드에 질소를 가수분해하는 야금성이다.
- RCN + HO2 → RC(O)NH2
이 효소들은 아크릴아미드를 생산하는데 상업적으로 사용된다.
아미드에 대한 질산염의 "무수 수화"는 옥소임을 수원으로 사용하여 입증되었다.[29]
- RCN + R'C(H)=노이 → RC(O)NH2 + R'CN
축소
질산은 다양한 금속 촉매를 통해 수소에 취약하다. 반응은 조건에 따라 1차 아민(RCHNH22) 또는 3차 아민(RCH2)3N 중 하나를 공급할 수 있다.[30] 기존의 유기 감소에서 질산은 아민에 대한 리튬 알루미늄 하이드라이드 처리로 감소한다. 알데히드로 가수분해된 후 이미인으로 환원되는 것은 스테판 알데히드 합성에서 일어나는데, 스테판 알데히드 합성에서는 산성 속에 스탠노스 염화물을 사용한다.
알킬화
알킬 질산은 니트리올 음이온을 형성하기에 충분한 산성이다. 음이온은 다양한 전기생성을 알킬 수 있다.[31] 예외적인 핵소독성의 핵심은 CN 유닛의 유도 안정화와 결합된 작은 강직 수요다. 이러한 특징들은 질산염을 약용 화학 합성에 사용하기 위해 강직적으로 까다로운 환경에서 새로운 탄소-탄소 결합을 만드는 데 이상적이다. 최근의 발전은 금속 반작용의 특성이 질소 질소나 인접한 핵탄소에 서로 다른 조율을 유발하며, 종종 반응성과 입체화학성의 차이가 심하다는 것을 보여주었다.[32]
뉴클레오필즈
니트릴의 탄소 중심은 전기영양성이므로 핵포화반응에 취약하다.
- 블라이즈 반응에 오르간오진 화합물로
- 피너 반응에 알코올이 들어있어
- 예를 들어, 아민 사코신과 시아나미드의 반응으로 크레아틴이[33] 생성된다.
- 질산은 후벤에서 프리델-크래프트 아틸화에서 반응한다.케톤에 대한 호이슈 반응
기타 방법 및 복합물
- 환원성 디시연화에서 니트릴 그룹은 양성자로 대체된다.[34] 금속 감소제(예: 테르트부탄올에 HMPA와 칼륨금속)를 용해하거나 KOH에 니트리올을 융합하여 해독할 수 있다.[35] 이와 유사하게 α-아미노니트라일은 리튬 알루미늄 하이드라이드와 같은 다른 환원제와 해독할 수 있다.[34]
- 핵포함에서 소르페 반응에 염기성 있는 질소성 자가 반응
- 유기농 화학에서 질산은 알키네에 카르보시아네이션에 첨가되는 것으로 알려져 있다.[36]

복잡화
질산은 시약과 촉매인 금속 니트리올 복합체의 전구체다. 예로는 [Cu(MecN)]4+와 PdCl2(PhCN)2[37]이 있다.
2.jpg)
니트릴 유도체
유기 시나미드
Cyamanide는 일반 구조 RRN-CN을 가진 N-cyano 화합물이며 무기체 모체 Cyamanide와 관련이 있다.
니트리올산화물
니트리올 산화물은 일반적인 구조 R-CNO를 가지고 있다.

발생 및 적용
질산은 다양한 식물과 동물원에서 자연적으로 발생한다. 120개 이상의 자연적으로 발생하는 질산은 지구와 해양으로부터 격리되었다. 질산은 과일 구덩이, 특히 아몬드에서 흔히 만나며, 수산화물을 통해 질소를 방출하는 브라시카 작물(양배추, 무두순, 콜리플라워 등)을 조리하는 과정에서 만난다. 아몬드나 일부 과일 구덩이를 섭취해 생산되는 시아노 무수인 만델로니트릴은 시안화수소를 방출하고 청록색성 글리코시드의 독성을 담당한다.[38]
30개 이상의 니트리올 함유 의약품은 현재 임상 개발에서 20개 이상의 니트리올 함유 리드와 함께 다양한 의약품 적응증에 대해 시판되고 있다. 니트릴 그룹은 상당히 튼튼하며 대부분의 경우 쉽게 신진대사를 하지 않고 그대로 신체를 통과한다.[citation needed] 질산염이 함유된 의약품의 종류는 당뇨병 치료제인 빌다글립틴부터 유방암 치료의 금본위제인 아나스트로졸까지 다양하다. 많은 경우 니트리올은 효소에 대한 기판에 존재하는 기능을 모방하는 반면, 다른 경우 니트리올은 물의 용해도를 증가시키거나 간에서 산화적 신진대사에 대한 민감도를 감소시킨다.[39] 니트리얼 기능성 그룹은 여러 가지 약품에서 발견된다.

선택적 세로토닌 재흡수 억제제(SSRI) 등급의 항우울제인 시탈로프람의 구조.





참고 항목
참조
- ^ IUPAC 골드 북 질산염
- ^ NCBI-MesH 니트릴레스
- ^ Karakida, Ken-ichi; Fukuyama, Tsutomu; Kuchitsu, Kozo (1974). "Molecular Structures of Hydrogen Cyanide and Acetonitrile as Studied by Gas Electron Diffraction". Bulletin of the Chemical Society of Japan. 47 (2): 299–304. doi:10.1246/bcsj.47.299.
- ^ 참조:
- Carl W. Scheele (1782) "Försök, beträffande det färgande ämnet uti Berlinerblå" (Experiment concerning the colored substance in Berlin blue), Kungliga Svenska Vetenskapsakademiens handlingar (Royal Swedish Academy of Science's Proceedings), 3: 264–275 (in Swedish).
- 라틴어로 재인쇄: 에른스트 벤자민 고틀립 헤벤스트리트(ed.)와 고트프리드 하인리히 셰퍼(트랜스), 오푸슐라 케미카 에 피르티카(Leipzig("Lipsiae")), (독일)와 함께 칼 빌헬름 스크렐레: 요한 고드프리드 뮐러, 1789), 제2권 148-174쪽.
- ^ David T. Mowry (1948). "The Preparation of Nitriles" (– Scholar search). Chemical Reviews. 42 (2): 189–283. doi:10.1021/cr60132a001. PMID 18914000.
{{cite journal}}: 외부 링크 위치format= - ^ 게이 루삭은 다음과 같이 순수하고 액화 수소 시안화수소를 생산했다.
- ^ J. Pelouze (1834). "Notiz über einen neuen Cyanäther" [Note on a new cyano-ether]. Annalen der Pharmacie. 10 (3): 249. doi:10.1002/jlac.18340100302.
- ^ 헤르만 Fehling(1844년)."Ueber Zersetzung 데 benzoësauren Ammoniaks 암모늄 벤조 산의 열에 의해 분해하는 것(탭에서 Wärme 죽durch 홀로 죽었다".Annalen하는 Chemie.doi:10.1002/jlac.18440490106 Pharmacie.49(1):91–97 und.이후 로랑은 그에 의해"nitrobenzoyl"–도"azobenzoyl"이미 exis 발견된 물질이라는 페이지 96일, Fehling:("다 로랑 소굴 폰 ihm entdeckten 몸 schon Nitrobenzoyl genannt 모자, Azobenzoyl existirt einauch schon, 그렇게könnte 남자 소굴 aus benzoësaurem Ammoniak entstehenden 몸 vielleicht Benzonitril nennen." 쓴다.ts – 그래서 벤조네이트 암모늄에서 유래한 물질의 이름을 "벤조니트릴"로 명명할 수 있을 것이다.
- ^ Pollak, Peter; Romeder, Gérard; Hagedorn, Ferdinand; Gelbke, Heinz-Peter (2000). Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a17_363.
- ^ Gregory, Robert J. H. (1999). "Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications". Chemical Reviews. 99 (12): 3649–3682. doi:10.1021/cr9902906. PMID 11849033.
- ^ "ISOBUTYRONITRILE". Organic Syntheses. 25: 61. 1945. doi:10.15227/orgsyn.025.0061.
- ^ "2-ETHYLHEXANONITRILE". Organic Syntheses. 32: 65. 1952. doi:10.15227/orgsyn.032.0065.
- ^ Chill, Samuel T.; Mebane, Robert C. (18 September 2009). "A Facile One-Pot Conversion of Aldehydes into Nitriles". Synthetic Communications. 39 (20): 3601–3606. doi:10.1080/00397910902788174.
- ^ C. Fizet; J. Streith (1974). "Hydroxylamine-O-sulfonic acid: A convenient reagent for the oxidative conversion of aldehydes into nitriles". Tetrahedron Lett. (in German). 15 (36): 3187–3188. doi:10.1016/S0040-4039(01)91857-X.
- ^ "o-Tolunitrile and p-Tolunitrile" H. T. Clarke and R. R. Read Org. 1941년 신스, 콜 제1권 514호
- ^ W. Nagata and M. Yoshioka (1988). "Diethylaluminum cyanide". Organic Syntheses.; Collective Volume, vol. 6, p. 436
- ^ W. Nagata, M. Yoshioka, and M. Murakami (1988). "Preparation of cyano compounds using alkylaluminum intermediates: 1-cyano-6-methoxy-3,4-dihydronaphthalene". Organic Syntheses.
{{cite journal}}: CS1 maint : 복수이름 : 저자목록(링크); - ^ Reynold C. Fuson; Oscar R. Kreimeier & Gilbert L. Nimmo (1930). "Ring Closures in the Cyclobutane Series. II. Cyclization Of α,α′-Dibromo-Adipic Esters". J. Am. Chem. Soc. 52 (10): 4074–4076. doi:10.1021/ja01373a046.
- ^ A. P. N. Franchimont (1872). "Ueber die Dibenzyldicarbonsäure" [On 2,3-diphenylsuccinic acid]. Berichte der Deutschen Chemischen Gesellschaft. 5 (2): 1048–1050. doi:10.1002/cber.187200502138.
- ^ J. Houben, Walter Fischer (1930) "Über eine neue Methode zur Darstellung cyclischer Nitrile durch katalytischen Abbau (I. Mitteil.)," Berichte der deutschen chemischen Gesellschaft (A and B Series) 63 (9): 2464 – 2472. doi:10.1002/cber.19300630920
- ^ Yamazaki, Shigekazu; Yamazaki, Yasuyuki (1990). "Nickel-catalyzed dehydrogenation of amines to nitriles". Bulletin of the Chemical Society of Japan. 63 (1): 301–303. doi:10.1246/bcsj.63.301.
- ^ Chen, Fen-Er; Kuang, Yun-Yan; Hui-Fang, Dai; Lu, Liang (2003). "A Selective and Mild Oxidation of Primary Amines to Nitriles with Trichloroisocyanuric Acid". Synthesis. 17 (17): 2629–2631. doi:10.1055/s-2003-42431.
- ^ Schäfer, H. J.; Feldhues, U. (1982). "Oxidation of Primary Aliphatic Amines to Nitriles at the Nickel Hydroxide Electrode". Synthesis. 1982 (2): 145–146. doi:10.1055/s-1982-29721.
- ^ Hiegel, Gene; Lewis, Justin; Bae, Jason (2004). "Conversion of α‐Amino Acids into Nitriles by Oxidative Decarboxylation with Trichloroisocyanuric Acid". Synthetic Communications. 34 (19): 3449–3453. doi:10.1081/SCC-200030958. S2CID 52208189.
- ^ Hampson, N; Lee, J; MacDonald, K (1972). "The oxidation of amino compounds at anodic silver". Electrochimica Acta. 17 (5): 921–955. doi:10.1016/0013-4686(72)90014-X.
- ^ Dakin, Henry Drysdale (1916). "The Oxidation of Amino-Acids to Cyanides". Biochemical Journal. 10 (2): 319–323. doi:10.1042/bj0100319. PMC 1258710. PMID 16742643.
- ^ Kukushkin, V. Yu.; Pombeiro, A. J. L. (2005). "Metal-mediated and metal-catalyzed hydrolysis of nitriles". Inorg. Chim. Acta. 358: 1–21. doi:10.1016/j.ica.2004.04.029.
- ^ Abbas, Khamis A. (1 January 2008). "Substituent Effects on the Hydrolysis of p-Substituted Benzonitriles in Sulfuric Acid Solutions at (25.0± 0.1) °C". Zeitschrift für Naturforschung A. 63 (9): 603–608. Bibcode:2008ZNatA..63..603A. doi:10.1515/zna-2008-0912. ISSN 1865-7109.
- ^ Dahye Kang, Jinwoo Lee, Hee-Yoon Lee (2012). "Anhydrous Hydration of Nitriles to Amides: p-Carbomethoxybenzamide". Organic Syntheses. 89: 66. doi:10.15227/orgsyn.089.0066.
{{cite journal}}: CS1 maint: 작성자 매개변수 사용(링크) - ^ Barrault, J.; Pouilloux, Y. (1997). "Catalytic Amination Reactions: Synthesis of fatty amines. Selectivity control in presence of multifunctional catalysts". Catalysis Today. 1997 (2): 137–153. doi:10.1016/S0920-5861(97)00006-0.
- ^ Adams, Roger (1957). Organic Reactions, Volume 9. New York: John Wiley & Sons, Inc. ISBN 9780471007265. Retrieved 18 July 2014.
- ^ Fleming, Fraser F.; Zhang, Zhiyu (24 January 2005). "Cyclic nitriles: tactical advantages in synthesis". Tetrahedron. 61 (4): 747–789. doi:10.1016/j.tet.2004.11.012.
- ^ Smith, Andri L.; Tan, Paula (2006). "Creatine Synthesis: An Undergraduate Organic Chemistry Laboratory Experiment". J. Chem. Educ. 83 (11): 1654. Bibcode:2006JChEd..83.1654S. doi:10.1021/ed083p1654.
- ^ a b 환원성 데시네이션 반응: 화학적 방법 및 합성 응용법 Jean-Marc Mattalia, Caroline Marchi-Delapierre, Hassan Hazimeh, Michel Chanon Arkivoc (AL-1755FR) pp 90–118 2006년 조항[permanent dead link]
- ^ Berkoff, Charles E.; Rivard, Donald E.; Kirkpatrick, David; Ives, Jeffrey L. (1980). "The Reductive Decyanation of Nitriles by Alkali Fusion". Synthetic Communications. 10 (12): 939–945. doi:10.1080/00397918008061855.
- ^ Yoshiaki Nakao; Akira Yada; Shiro Ebata & Tamejiro Hiyama (2007). "A Dramatic Effect of Lewis-Acid Catalysts on Nickel-Catalyzed Carbocyanation of Alkynes". J. Am. Chem. Soc. (Communication). 129 (9): 2428–2429. doi:10.1021/ja067364x. PMID 17295484.
{{cite journal}}:format=필요로 하다url=(도움말) - ^ Rach, S. F.; Kühn, F. E. (2009). "Nitrile Ligated Transition Metal Complexes with Weakly Coordinating Counteranions and Their Catalytic Applications". Chemical Reviews. 109 (5): 2061–2080. doi:10.1021/cr800270h. PMID 19326858.
- ^ 천연물 보고서 제 5, 1999 Nitrile 함유 천연물
- ^ Fleming, Fraser F.; Yao, Lihua; Ravikumar, P. C.; Funk, Lee; Shook, Brian C. (November 2010). "Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore". J Med Chem. 53 (22): 7902–17. doi:10.1021/jm100762r. PMC 2988972. PMID 20804202.



{kind=link}