1,3-다극성 사이클로어 추가
1,3-Dipolar cycloaddition| 후이젠 1,3-다극 사이클로어데이션 | |
|---|---|
| 이름을 따서 명명됨 | 롤프 위스겐 |
| 반응형 | 링 형성 반응 |
| 식별자 | |
| 유기화학포털 | 후이젠-1,3극자-사이클로더덕션 |
| RSC 온톨로지 ID | RXNO:0000018 |
1,3극 사이클로어데이션은 1,3극과 이극성 쌍극성 간 화학반응으로 5극성 고리를 형성한다. 초기 1,3극 사이클로아드는 1,3극이 발견된 후 19세기 후반에서 20세기 초반에 묘사되었다. 기계론적 조사와 합성 적용은 주로 롤프 휘스겐의 작업을 통해 1960년대에 확립되었다.[1] 따라서, 그 반응을 Huisgen cycloaddition이라고 부르기도 한다(이 용어는 종종 1,2,3-triazole을 생성하기 위한 유기 아지드와 알키네 사이의 1,3-dippolar cycloaddition을 구체적으로 묘사하기 위해 사용된다). 1,3-dippolar cycloaddition은 5-mbles와 tte의 섭생성격의 중요한 합성 경로다.상속인 고리개봉형 고리개봉형 고리유형 파생상품 이극성 소립자는 전형적으로 알켄이나 알킨이지만 다른 파이 계통이 될 수 있다. 2극성애호가 알키네일 때는 일반적으로 방향족 고리가 생산된다.

기계론적 개요
원래 제안된 두 개의 메커니즘은 1,3-다극성 사이클로어 추가에 대해 기술한다. 첫째, 롤프 후이젠이 제안한 결합형 사이클로 추가 메커니즘과 [2]둘째, 파이어스톤이 제안한 디라디컬 중간을 포함하는 단계별 메커니즘이다.[3] 많은 논쟁을 거친 후, 이전의 제안은 현재 일반적으로[4] 받아들여지고 있다. 즉 1,3-디폴은 결합되고, 종종 비동기적이며, 대칭성이 허용되는 4+2ss 패션으로 6-전자의 Huckel 방향 전환 상태를 통해 2-폴라로피와 반응한다. 단, 촉매가 없는 1,3극 사이클로이드와 [5]니트리올 산화물의[6] 반응에 대한 몇 가지 단계적 메커니즘의 예가 존재한다.

순환 메커니즘
Huisgen은 1,3-dipolar diazo 화합물과 다양한 2극성 알케인 사이의 일련의 사이클로아드를 조사했다.[2] 다음의 관찰은 일치된 순환 순환 메커니즘을 지지하며, 단계적 비행 경로나 단계적 극지 경로를 반박한다.
- 대체 효과: 쌍극자의 다른 대체물은 사이클로 추가율에 큰 영향을 주지 않으며, 이는 반응이 충전 분리 중간을 포함하지 않음을 시사한다.
- 용제 효과: 용제 극성은 반응제에서 전환 상태로 가는 과정에서 극성이 크게 변하지 않는 순환기 메커니즘에 따라 사이클로 추가 속도에 거의 영향을 미치지 않는다.
- 입체화학: 1,3-다극성 사이클로아데이션은 2극성 쌍극성(즉, 동기생산을 주는 시스알케네)과 관련하여 항상 입체적인 것으로, 두 시그마 결합이 동시에 형성되는 일치된 순환 순환 메커니즘을 지원한다.
- 열역학 매개변수: 1,3극성 사이클로아드는 디엘-알데르 반응과 유사한 활성화의 음 엔트로피가 비정상적으로 크므로 전환 상태가 고도로 순서가 지정됨을 시사하며, 이는 결합된 순환 순환 반응의 시그니처다.
1,3-디폴

1,3-디폴은 아릴형 또는 프로파릴/알레닐형 zwitterionic octet/sextet 구조로 나타낼 수 있는 유기 분자다. 두 종류의 1,3-디폴은 모두 3개의 원자에 걸쳐 π-시스템에서 4개의 전자를 공유한다. 아라이엘형은 구부러진 반면 프로파릴/알레닐형은 기하학적으로 선형적이다.[7] 유황이나 인과 같은 고열원소를 함유한 1,3-디폴도 알려져 있지만 일상적으로 덜 활용된다.
공명 구조는 1,3-디폴의 모든 종단부에 음전하와 양의 전하를 모두 분산시키기 위해 그릴 수 있다(아래 방식 참조). 1,3-디폴의 전자분포를 설명하는 보다 정확한 방법은 쌍극자 모멘트 측정이나[8] 계산과 같은 실험 또는 이론적 데이터를 기반으로 주요 공명 기여자를 할당하는 것이다.[9] 예를 들어, 디아조메탄은 단자 질소 원자에서 가장 큰 음성을 가지고 있는 반면, 히드라조산은 내부 질소 원자에서 가장 큰 음성을 가지고 있다.

결과적으로, 이 양면성은 1,3-디폴의 끝이 동시에 핵종양과 전기생식으로 처리될 수 있다는 것을 의미한다. 각 끝에서 핵소독성과 전기소독성의 정도는 프런티어 분자 궤도를 이용하여 평가할 수 있으며, 이는 계산적으로 얻을 수 있다. 일반적으로, HOMO에서 가장 큰 궤도 계수를 가진 원자는 핵분열체 역할을 하는 반면, LUMO에서는 핵분열체 역할을 한다. 가장 많은 핵종 원자는 보통, 그러나 항상은 아니지만, 가장 많은 전자가 풍부한 원자가 된다.[10][11][12] 1,3-다극성 사이클로아드화에서 쌍극성-다극성 쌍의 정체성은 1,3-디폴로의 HOMO 또는 LUMO 캐릭터가 지배할지 여부를 결정한다(아래 분자 궤도에 대한 논의 참조).
쌍극성체
가장 일반적으로 사용되는 이극성애자는 알케인과 알케인이다. 카보닐이나 이미인과 같은 이질 원자성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질성 이질체성 2극성질체의 다른 예로는 풀레네와 나노튜브가 있는데, 프라토 반응에서 아조메틴 이라이드와 함께 1,3극성 사이클로어데이션(Cycloaddition)을 겪을 수 있다.
용제 효과
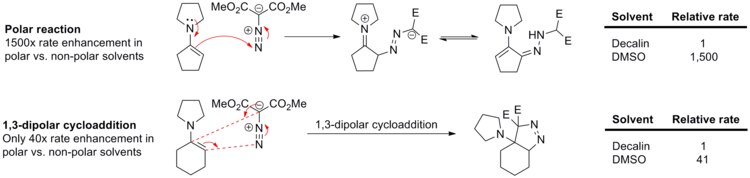
1,3극 사이클로아데이션은 반응물질과 전이상태 모두 일반적으로 비흡수성이기 때문에 용매효과를 거의 경험하지 않는다. 예를 들어 페닐 디아조메탄과 에틸 아크릴레이트 또는 노르보르네(아래 방식 참조)[13] 사이의 반응 속도는 사이클로헥산에서 메탄올에 이르는 다양한 용매에 의해 약간만 변화한다.

1,3-dipolar cycloaddition에서의 용제 효과의 부족은 에나민과 디메틸 디아조말론산염의 반응에서 명확히 입증된다(아래 방법 참조).[14] 디아조 화합물에 N-사이클로펜테닐 피롤리딘 핵포틸이 첨가된 극반응은 비극성 데칼린보다 극성 DMSO에서 1,500배 더 빠르게 진행된다. 반면, 이러한 반응의 밀접한 유사점인 디메틸 디아조말론산염에 대한 N-사이클로헥세닐 피롤리딘 1,3-다극성 사이클로아데스는 데칼린에 비해 DMSO에서 41배밖에 속도를 내지 못한다.
프런티어 분자 궤도 이론
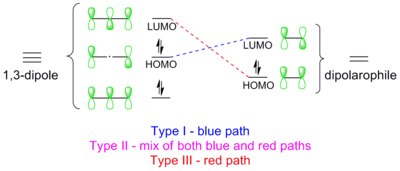
1,3-다극성 사이클로아드는 드워-지머만 규칙과 우드워드-호프만 규칙을 따르는 순환 반응이다. 드워-지머만 치료에서, 반응은 이 특정한 분자 궤도 다이어그램에 대한 5-중앙, 0-노드, 6-전자 허클 전환 상태를 통해 진행된다. 그러나 각 궤도에는 같은 결과에 도달하기 위한 기호가 무작위로 할당될 수 있다. 우드워드-호프만 치료에서는 1,3-디폴의 프런티어 분자 궤도(FMO)와 2극성 분자 궤도(FMO)가 대칭 허용 4+2s 방식으로 겹친다s. 그러한 궤도 중첩은 타입 I, II, III의 세 가지 방법으로 달성될 수 있다.[15] 지배적인 통로는 가장 작은 HOMO-LUMO 에너지 격차를 가지고 있는 통로다.
제1종
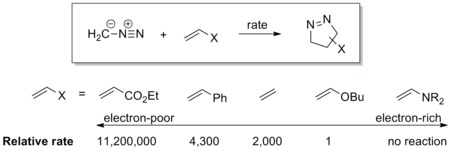
쌍극자에는 쌍극자 LUMO와 겹치는 고화질 HOMO가 있다. 이 등급의 쌍극자는 호모 제어 쌍극체 또는 핵성 쌍극체로 불리며, 여기에는 아조메틴 이라이드, 카르보닐 이라이드, 니트리올 이라이드, 아조메틸 이미인, 카보닐 이미인, 디아조알카인이 포함된다. 이 쌍극점들은 전기영양성을 쉽게 증가시킨다. 2극성질체 위의 전자-배출 그룹(EWG)은 LUMO를 낮춰 반응을 가속하는 반면, 전자 기부 그룹(EDG)은 HOMO를 상승시켜 반응을 감속시킨다. 예를 들어, 일련의 2극성질체에 대한 디아조메탄의 반응성 척도는 아래 계획에 나와 있다. 디아조메탄은 전자 빈약 에틸 아크릴산 에틸과 전자 부틸 비닐에테르보다 100만 배 이상 빠르게 반응한다.[16]
이 유형은 디엔 호모(Diene HOMO)와 디엔 호모(Diene HOMO)가 디엔호메(Diene HOMO)와 결합하는 일반적인 전자 수요 디엘-알데르 반응과 닮았다.
타입 II
쌍극체의 HOMO는 쌍극체의 LUMO와 결합할 수 있고, 또는 쌍극체의 HOMO는 쌍극체의 LUMO와 결합할 수 있다. 이러한 양방향 상호작용은 양쪽 방향의 에너지 갭이 유사하기 때문에 발생한다. 이 등급의 쌍극자를 호모-LUMO 제어 쌍극자 또는 암비필 쌍극자(Nitrile imide, nitrone, carbonyl oxide, nitrile oxide, azide)라고 한다. 양극성질 대체물은 상호작용하는 두 궤도 사이의 에너지 격차를 낮춰 반응을 가속화할 수 있다. 즉, EWG는 LUMO를 낮추는 반면 EDG는 HOMO를 상승시킨다. 예를 들어, 아지드화물은 유사한 반응성을 가진 다양한 전자 풍부하고 전자가 부족한 양극성질체와 반응한다(아래 반응도 척도 참조).[17]

타입 III
쌍극자(dipole)는 2극자성(dipolarophile)의 HOMO와 중첩되는 저층 LUMO를 가지고 있다(도표에서 빨간 점선으로 표시됨). 이 등급의 쌍극자는 아산화질소와 오존을 포함하는 LUMO 제어 쌍극형 또는 전기영양 쌍극형이라고 한다. 이중극성애세포의 EWG는 반응을 감속하고 EDG는 반응을 가속화한다. 예를 들어, 오존은 전자가 풍부한 2-메틸프로펜과 전자가 빈약한 테트라클로로메테인보다 약 10만배 빠르게 반응한다(아래 반응도 척도 참조).[18]
이 타입은 디엔 루모가 디에노필 HOMO와 결합하는 역전자 수요 디엘스-알더 반응과 닮았다.

반응도
1,3 사이클 추가와 같은 결합 공정은 고도로 순서가 정해진 전환 상태(활성화의 음성 엔트로피)를 필요로 하며 중간 정도의 엔탈피 요건만 필요로 한다. 경기 반응 실험을 사용하여, 다른 사이클로 추가 반응에 대한 상대적인 추가 비율이 반응성의 요인에 대한 일반적인 결과를 제공하는 것으로 밝혀졌다.
- 특히 방향족과 함께 결합하면 전환 상태의 안정화에 의한 반응 속도가 증가한다. 전환 중, 두 시그마 결합은 서로 다른 비율로 형성되고 있으며, 이는 결합 대체물로 전하 분배를 통해 안정화될 수 있는 전환 상태에서 부분적인 전하를 발생시킬 수 있다.
- 분산 전자 구름이 전자의 흐름을 시작하는 데 더 적합하기 때문에 더 많은 편극성 이극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성은 더 반응적이다.
- 지반 상태의 에너지 증가로 인해 각진 균형이 높은 이극성질소는 반응성이 더 높다.
- 방해받지 않는 반응물질의 결과로 전환 상태에서 강한 방해물이 증가하면 반응률이 급격히 낮아진다.
- 이질-이질-이질-이질-이질-이질-이질-이질-이질-이질-이질-이질-이질-이질-이질-은 전환 상태 중 파이 결합 손실을 상쇄하기 위해 시그마 결합 에너지의 낮은 이득으로 인해 C,C-이질-이질-이질-이질체보다 더 느리게 첨가된다.
- 2극성 이소체성(trans-stille imide)은 스테로닉에 의한 반응율에 영향을 미친다. 트랜스 이소체는 반응하는 동안 120°의 결합 각도가 109°로 줄어들어 강직성 충돌 증대를 위해 서로 쪽으로 시스 대체물을 흐리게 하기 때문에 더 반응한다(trans-stille imide는 시스틸혜보다 27배 더 빠르다).

입체도
1,3극성 사이클로아드는 일반적으로 1,3극성 및 2극성 사이클로필드에 대한 구성을 유지하게 된다. 그러한 높은 수준의 입체감은 단계적 반응 메커니즘을 통해 결합되는 강한 지지력이다. 앞서 언급했듯이, 많은 예들은 반응이 단계적이었음을 보여주며, 따라서 부분적인 입체감을 나타내거나 전혀 보이지 않는다.
쌍극성애자(dipololarophile)와 관련하여
이극성 알켄의 cis-대체물질은 cis가 되고, 그 결과 5-membed cycronic cycle complex(아래 방법 참조)[19]에서 transfer가 된다.

쌍극자 관련
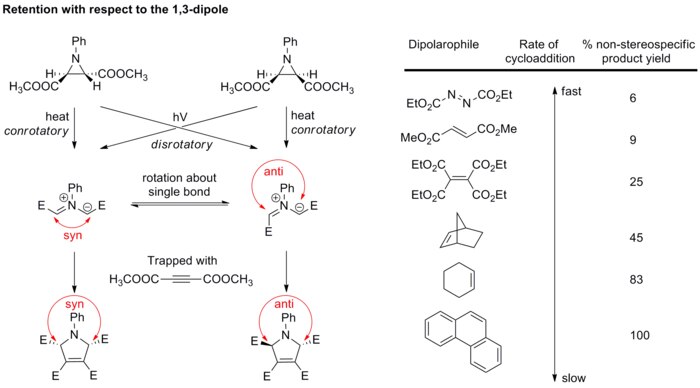
일반적으로 쌍극체의 입체화학은 극소수의 이중극만이 입체적인 중심을 형성할 수 있고 공명 구조는 입체화학을 교란시키는 본드 회전을 허용하기 때문에 큰 문제가 되지 않는다. 그러나 아조메틴 이라이데스의 연구는 사이클로어데이션이 쌍극성분에 관해서도 입체적이라는 것을 입증했다. 디아스트레오퓨어 아조메틴 이라이드는 아지리딘의 전기 순환 고리 개방에 의해 생성된 다음 접합 회전이 일어나기 전에 강한 이극성 섬유로 빠르게 갇힌다(아래 구성 참조).[20][21] 약한 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극
이러한 결과는 1,3극 사이클로어디가 입체적이라는 것을 모두 확인시켜 1,3극 사이클로어드와 2극성 사이클로필드를 모두 보존할 수 있게 한다.
탈지극성
반응하는 동안 두 개 이상의 스테레오센서가 생성되면 이질화 전환 상태와 제품을 얻을 수 있다. Diels-Alder cycloaddition에서는 보통 2차 궤도 상호작용에 의한 내분비역회전율이 관찰된다. 그러나 1,3극 사이클로어드화에서 두 가지 힘은 이뇨반응성에 영향을 미친다: 매력적인 π-상호작용(Diels-Alder Cycloadation에서 이차 궤도 상호작용 포함)과 혐오스러운 steric 상호작용이다. 불행하게도, 이 두 힘은 종종 서로를 취소하여 1,3극 사이클로어 추가에서 좋지 않은 이질제거를 유발한다.
기질 제어 이질제거제 1,3-다극 사이클로아드의 예는 다음과 같다. 첫째는 벤조니트릴 N벤질라이드와 메틸 아크릴레이트 사이의 반응이다. 전환 상태에서는 페닐과 메틸에스테르 그룹이 쌓여서 시스 하위체를 배타적 최종 피로라인 제품으로 준다. 이 호의적인 π-상호작용은 페닐과 메틸에스테르군 사이의 강직성 거부반응을 상쇄시킨다.[22] 두 번째는 니트로네와 디하이드로푸란의 반응이다. 엑소 선택성은 강한 거부감을 최소화하기 위해 달성된다.[23] 마지막으로 알켄에 대한 근육내 아조메틴 이라이드 반응이다. 이질제거성은 덜 긴장된 시스 퓨즈 링 시스템의 형성에 의해 제어된다.[24]

방향 1,3극 사이클로어 추가
사이클로어데이션의 궤적을 조절하여 이질제거 반응을 얻을 수 있다. 예를 들어, 금속은 2극성 쌍극체와 들어오는 쌍극자까지 킬레이트하여 한 면에 선택적으로 사이클로어데이션(Cycloaddition)을 지시할 수 있다. 아래의 예는 마그네슘 이온이 존재하는 상태에서 산화질소를 항산화 순수 아릴 알코올에 첨가하는 것을 보여준다. 알켄의 가장 안정적인 순응은 히드록실 그룹을 알켄의 평면 위에 놓는다. 그리고 나서 마그네슘은 히드록실 그룹과 질소산화물의 산소 원자까지 킬레이트한다. 따라서 시클로데이션은 상단 얼굴에서 선택적으로 나온다.[25]
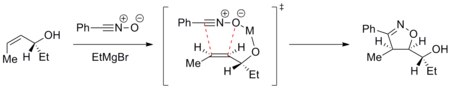

섭정률
비대칭 쌍극 쌍극 쌍의 경우 두 개의 레지오이소메릭 제품이 가능하다. 전자/스테레오전자 및 강체 인자는 모두 1,3극 사이클로아드의 섭정율에 기여한다.[27]
전자/철전자 효과
지배적인 전자적 상호작용은 가장 큰 HOMO와 가장 큰 LUMO의 조합이다. 따라서, 가장 큰 궤도 HOMO와 LUMO 계수를 갖는 원자에 의해 섭생성이 지배된다.[28][29]
예를 들어, 세 가지 이중극성 물질(메틸 아크릴레이트, 스티렌 또는 메틸 시나몬산)에 대한 디아조메탄의 사이클로 추가를 고려하십시오. 디아조메탄의 탄소는 HOMO가 가장 큰 반면, 메틸 아크릴레이트 및 스티렌의 엔드 올레피닌 탄소는 가장 큰 LUMO를 가지고 있다. 따라서 사이클로어데이션은 C-3 위치에서 역선택적으로 대체한다. 시나몬 메틸의 경우, 두 대체물(Ph v.s. COOMe)이 알켄에서 전자를 추출하기 위해 경쟁한다. 카복실(carboxyl)은 더 나은 전자-탈출 그룹으로서 β-탄소를 가장 전기적으로 만드는 그룹이다. 따라서 사이클로어데이션은 C-3에서 카복실 그룹을, C-4에서는 페닐 그룹을 섭생적으로 산출한다.

스테리크 효과
스테릭 효과는 앞서 언급한 전자 효과와 협력하거나 경쟁할 수 있다. 때로는 강한 효과가 전자적 선호도를 완전히 능가하여 반대편 레지오이소머만을 독점적으로 부여한다.[30]
예를 들어, 디아조메탄은 일반적으로 3-카르복실 피라졸린을 주기 위해 아크릴산 메틸에 첨가된다. 하지만, 시스템에 더 강한 요구를 넣음으로써, 우리는 이소메릭 4-카르복실 피라졸린을 관찰하기 시작한다. 이 두 레지오이소머의 비율은 견고한 수요에 달려 있다. 극단적으로, 수소에서 t-부틸로 크기를 증가시키면 섭생성이 100% 3-카르복실 대체가 100%에서 100% 4-카르복실 대체가 된다.[31][32]

합성 응용 프로그램
1,3극성 사이클로아데이션은 3각형, 후란형, 이소사졸, 피롤리딘 등과 같은 많은 중요한 5각형 이성계의 합성을 위한 중요한 방법이다. 또한 일부 사이클로아덕트는 선형 골격을 나타내기 위해 분할할 수 있으며, 이는 알리프 화합물의 합성을 위한 또 다른 경로를 제공한다. 이러한 반응들은 입체적이고, 이질적이며, 재생적이기 때문에 또한 엄청나게 유용하다. 아래에 몇 가지 예가 제시되어 있다.
니트리올산화물
질화산화물을 함유한 1,3극 사이클로어데이션은 널리 사용되는 복면-알돌 반응이다. 아산화질소와 알켄 사이의 사이클로어드는 순환 이소사졸린 제품을 산출하는 반면 알킨과의 반응은 이소사졸을 산출한다. 이소사졸린과 이소사졸은 모두 수소화에 의해 분해되어 각각 알돌형 β-히드록시카르보닐 또는 클라이센형 β-디카르보닐 제품을 드러낼 수 있다.
아래 그림과 같이 미야콜라이드의 합성에는 아산화질소-알킨 사이클로어데이션에 이은 아산화질소-알킨 사이클로어데이션이 활용되었다.[33]

카보닐 이라이드
1,3극성 사이클로어드 추가 반응은 의약, 생물학, 기계학 연구를 위한 복잡한 순환 계통과 분자의 합성에서 강력한 도구로 떠올랐다. 그 중 산소를 함유한 5-membed 순환분자를 생성하기 위해 카보닐 일라이드를 포함하는 [3+2] 사이클로어드 반응이 광범위하게 채택되었다.[34]
1,3-다극 사이클로어 추가반응을 위한 카보닐 이라이드 준비
이라이드는 음전하 탄소 원자에 연결된 양전하 이성질체로 간주되며, 여기에는 이라이드(sulfium), 티오카르보닐(tiocarbonyl), 옥소늄(oxium), 질소(gene), 카보닐(carbonyl)이 포함된다.[35] [3+2] 사이클로어드 추가 반응을 위해 산소가 함유된 5-엠베드 링 구조를 생성하는 데 필요한 매개체인 카보닐 이라이드를 생성하기 위한 몇 가지 방법이 존재한다.
광촉매에 의한 디아조메탄 유도체의 카르보닐화합성
카보닐 이라이드 합성의 가장 초기 사례 중 하나는 광투석을 포함한다.[36] 테트라메틸루레아가 있는 상태에서 디아조테트라키스(trifluoromethyl)사이클로펜타디엔*(DTTC)을 광분해하면 분자간 핵소독성 공격과 그에 따른 DTTC 모이티 아로마화에 의해 카보닐 닐라이드를 생성할 수 있다.[36] 이것은 방향성에 의해 전달되는 안정성, 전자에서 트라이플루오로메틸 그룹을 철수시키고 전자에서 디메틸아민 그룹을 기증하기 때문에 X선 결정술로 분리되고 특징지어졌다. 안정적 카보닐 이라이드 디폴레는 이극성질체와의 [3+2] 사이클로 추가 반응에 사용될 수 있다.

광투석에 의한 카보닐 이라이드 합성의 또 다른 초기 예는 Olah 등이 보고하였다.[37] 디데터리오디아조메탄은 포름알데히드가 있는 곳에서 광학화하여 디데터리오폼알데히드 카르보닐 이라이드를 생성하였다.

양성자전달에 의한 히드록시피로네에서 카르보닐 이라이드의 합성
카보닐 이라이드는 금속 촉매가 없을 때 히드록시-3-피로네의 산성 촉매에 의해 합성될 수 있다.[38] 초기 tautomerization이 발생하고, 피론 링을 방향화하고 카보닐 ylide를 생성하기 위해 이탈 그룹이 제거된다. 2극성애기와 함께 사이클로어 추가 반응이 마지막으로 옥사시클을 형성한다. 이 접근방식은 제한된 효용과 피론 해골에 대한 요구사항 때문에 덜 널리 채택된다.

5-sexy-4-pyrones는 또한 분자 내 수소 전달에 의해 카보닐 이라이드를 합성하는데 사용될 수 있다.[39] 수소 전달 후 카보닐 이라이드는 2극성 물질과 반응하여 산소가 함유된 링을 형성할 수 있다.

디할로카르베네에서 α-할로카르보닐 일라이드 합성
디할로카베네스는 또한 디할로카베네스의 전자 인출 특성을 이용하여 카보닐 이라이드를 생성하기 위해 고용되었다.[40][41][42] 페닐(트리클로메틸)머큐리와 페닐(트리브로메틸)머큐리는 각각 디클로로카베네와 디브로모카베네이다. 카보닐 이라이드는 케톤이나 알데히드와의 디할로카르베네 반응에 의해 생성될 수 있다. 그러나 α-할로카르보닐 이라이데스의 합성은 또한 바람직하지 않게 일산화탄소의 손실과 탈산소 생성물의 생성을 초래할 수 있다.

금속 촉매에 의한 디아조메탄 유도체의 카르보닐 이라이드 합성
카보닐 이릴화물을 생성하기 위한 보편적 접근방식은 일반적으로 디코퍼 또는 디호듐 촉매가 존재하는 α-디아조카르보닐 화합물의 금속 카탈루션을 포함한다.[43] 질소 가스를 방출하고 금속으로 전환한 후 카보닐 그룹과의 분자간 반응으로 카보닐 일라이드가 생성될 수 있다. 알켄 또는 알킨 이극성 2극성 물질에 대한 후속 사이클로 추가 반응은 산소가 함유된 5흡성 링을 제공할 수 있다. 옥사시클 합성에 적당한 수율을 주는 촉매로는 Rh2(OAc)4와 Cu(acacc)가 있다.2[44][45]

디아조카르보닐 화합물의 금속촉매에 의해 매개된 1,3-다극성 사이클로어 추가반응의 메커니즘
산소가 함유된 5-흡입 고리를 합성하기 위해 디아조카르보닐 분자의 금속 강직으로 매개되는 1,3-다극성 사이클로어드 반응의 보편성과 광범위한 사용은 그 메커니즘에 상당한 관심을 불러일으켰다. 몇몇 그룹은 레지오 선택성과 스테레오 선택성과 관련하여 합성 분자의 범위를 확장하기 위한 메커니즘을 연구했다. 그러나 이러한 반응의 높은 턴오버 빈도 때문에 중간자와 메커니즘은 여전히 이해하기 어렵다. 일반적으로 받아들여지는 메커니즘은 안정적인 루테늄-카르브노이드 복합체와[46] 로듐 메탈로카르베네스의 특성화에 의해 개발되며,[47] 디아조 화합물에서 금속-카르브노이드 복합체가 초기 형성되는 것을 포함한다. 그러면 질소 가스의 제거는 야금 혜택을 준다. 카보닐 산소에 의한 분자내 핵소피질은 금속 촉매를 재생시켜 카보닐 닐라이드를 형성한다. 그런 다음 카보닐 이라이드는 산화칼을 생성하기 위해 디메틸 아세틸렌디카르복실산(DMAD)과 같은 알켄이나 알킨과 반응할 수 있다.

그러나 야금성 중간에서 카보닐 이라이드가 생성되는지는 불확실하다. 어떤 경우에, 야금류는 또한 2극성포체와 직접적으로 반응할 수 있다.[48] 이 경우 디르호듐(dirhodium)과 같은 야금 혜택(metalocarbene)이 있다.II)Tetracarboxylate carbene은 고주파 금속 에놀레이트 형태의 상호작용을 통해 안정화된다.[49][50][51][52] 후속 1,3극 사이클로어 추가 반응은 과도 금속 복합 카보닐 이라이드를 통해 발생한다. 따라서 지속적인 야금성은 금속 리간드의 입체화학과 크기에 기초한 1,3극 사이클로어 추가 반응의 입체적 선택성과 재생성에 영향을 미칠 수 있다.
tetracarboxylate_metallocarbene_stabilized_by_%CF%80C-Rh%E2%86%92%CF%80C%3DO_hyperconjugation..png)
카르보닐 이라이드 쌍극체와 알키닐 또는 알케닐 이극성 사이의 1,3-다극성 사이클로어 추가 반응의 메커니즘은 재열성 및 입체성과 관련하여 광범위하게 조사되었다. 대칭 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 2극성 [52]2 반대로 비대칭 2극성 2극성체는 복수의 레지오이저와 입체자를 가질 수 있다. 이러한 레지오이저와 스테레오이오머는 프런티어 분자 궤도(FMO) 이론, 스테레오 인터랙션 및 스테레오 전자 상호작용에 기초하여 예측될 수 있다.[53][54]

디아조카르보닐 화합물의 금속 촉매에 의해 매개된 1,3-다극성 사이클로어드 반응의 레지오셀레이션성
정의된 레지오케미컬로 분자를 생성하기 위해서는 카르보닐 이라이드 디폴로와 알키닐 또는 알케닐 이극성 디폴로필드의 1,3-디폴라드 반응의 레지오셀레이션이 필수적이다. FMO 이론과 쌍극성 간 HOMO-LUMO 에너지 갭 분석은 실험 결과의 섭정성을 합리화하고 예측할 수 있다.[55][56] HOMO와 LUMO는 이중극자 또는 이중극자 중 하나에 속할 수 있으며, 이 경우 HOMO-LUMO 또는 HOMO-LUMO 상호작용이 존재할 수 있다. 가장 큰 계수와 궤도의 중첩은 궁극적으로 결과를 합리화하고 예측할 수 있다.

파드와 동료들은 카보닐 이라이드 쌍극에 의해 매개된 1,3극성 사이클로어 추가 반응의 원형적 섭생성을 검사했다.[54][57] 벤젠에 Rh(OAC2)4 촉매를 이용해 디아조디온은 메틸 프로피올레이트, 메틸 프로파르길 에테르와 함께 1,3극 사이클로어 추가 반응을 보였다. 메틸 프로피올레이트와의 반응은 카보닐 yliide의 카보닐 그룹에 대한 탄소 근위부와 메틸 프로피올레이트 단자 알키인 탄소에 대한 가장 큰 계수를 갖는 HOMO-LUMO 상호작용에 따른 주요 계수를 가진 두 개의 레지오이소머를 제공한다. 메틸 프로파르길 에테르와의 반응은 카보닐 일라이드의 카보닐 그룹과 메틸 프로파길 에테르 단자 알킨 탄소에 대한 탄소 원위계수가 가장 큰 호모-LUMO 상호작용에 따른 하나의 레지오이소머를 제공한다.

디아조카르보닐 화합물의 금속 촉매에 의해 매개되는 1,3-다극성 사이클로어드 반응의 레지오셀레이션성은 또한 안정적인 야금성 형성을 통해 금속의 영향을 받을 수 있다.[48][58] 금속 에놀레이트 형태의 상호작용을 통해 야금류의 안정화는 카르보닐 이라이드의 형성을 방해하여 야금류 쌍극체와 알킬니엘 또는 알케닐 이극성질(Dirhodium:ⅡC-Rh)tetracarboxylate metalocarbene은 →→πC=O→π초과점화)에 의해 안정화된다. 이런 상황에서 금속 리간드는 1,3극 사이클로어 추가반응의 섭열성과 입체감에 영향을 줄 것이다.
디아조카르보닐 화합물의 금속 강직으로 매개된 1,3극 사이클로어드 추가 반응의 입체감각성 및 비대칭
카르보닐 이라이드 디폴로와 알케닐 이극성 디폴로필드의 1,3차원 시클로아드 반응의 입체성도 면밀하게 검토되었다. 알키닐 2극성 편성의 경우, 비교적 평탄한 탄소가2 형성되므로 입체감이 문제가 되지 않는 반면, 역극성은 반드시 고려되어야 한다(카보닐 Ylide Dipoles와 알케닐 또는 알키니 2극성 편광체 사이의 1,3극성 사이클로어 추가 반응 제품 이미지 참조). 단, 알케닐 이극성질체의 경우 제품종에서 탄소가3 생성되기 때문에 섭생성과 입체성 모두를 고려해야 한다.
카보닐 이라이드 디폴로와 알케닐 이극성 디폴로필드의 1,3극성 사이클로어 추가 반응은 이염성 제품을 생성할 수 있다.[52] exo 제품은 옥사시클의 에테르 다리에서 2극성 대체물이 시스인 것이 특징이다. 엔도 제품은 옥사시클의 에테르 다리로 2극성 대체물이 전달되는 것이 특징이다. 두 제품 모두 동기식 또는 비동기식 결합 프로세스를 포함하는 순환 전환 상태를 통해 생성될 수 있다.
한 초기 예는 금속 촉매와 루이스 산을 가진 엔도 및 엑소 제품 측면에서 입체감을 부여했다.[59] 금속촉매 Rh2(OAc)4만으로는 엑소 제품을, 추가 Lewis acid Yb(OTF)3로는 엔도 제품을 선호한다. Lewis acid cycloaddition reaction에 대해 관측된 엔도 선택성은 Yb(Otf)(3LUMO)가 조정한 이극성(Dipollophile)과 이극성(HOMO) 사이에 카보닐 π 시스템의 궤도 중첩이 최적화되었기 때문이다. 많은 조사 후 카보닐 ylide cycycycycycycycycycycycycycycycycycycycladd의 입체성에 영향을 미치는 두 가지 주요 접근방법이 있다.금속 촉매와 루이스 산의 치맛을 이용하는 것이 개발되었다.[52]


첫 번째 접근법은 치랄 금속 촉매를 사용하여 엔도와 엑소 입체감을 변조한다. 특히 Rh2[(S)-DOSP]4와 Rh2[(S)-BPTV]4는 중간 정도의 비대칭 유도를 유도할 수 있으며 항응고제 유사극산 A를 합성하는 데 사용되었다.[60] 이는 사이클로어데이션 동안 카보닐 일라이드와 관련된 치랄 금속 촉매가 남아 안면 선택성을 방해한 결과다. 그러나 정확한 메커니즘은 아직 완전히 이해되지 않았다.

두 번째 접근법은 치랄 루이스 산 촉매를 사용하여 아치랄 금속 촉매를 사용하여 카보닐 일라이드 생성 후 안면 입체감을 유도한다.[61] 치랄 루이스산 촉매는 이극성 촉매와 조응하여 이극성 촉매의 LUMO를 낮추는 동시에 항항우울증으로 이어지는 것으로 생각된다.

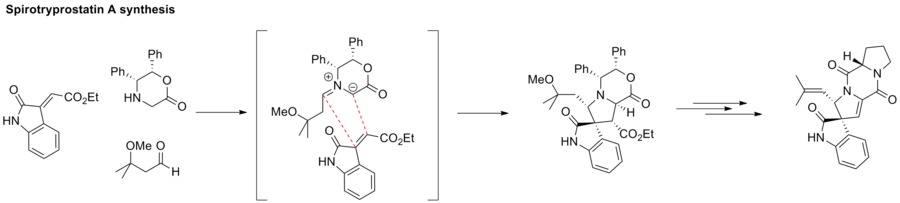
아조메틴 이라이데스
1,3-Azomethine ylide와 알켄 사이의 Dipolar cycloaddation은 피롤리딘과 같은 아지시클릭 구조를 제공한다. 이 전략은 스피로트리프로스타틴 A의 합성에 적용되었다.[62]
오존
오조놀리시스(Ozonolyis)는 매우 중요한 유기적 반응이다. 알케네와 알케인은 오조놀리시스(ozonolyis)에 의해 분해되어 알데히드, 케톤 또는 카르복실산 제품을 제공할 수 있다.
생물학적 응용
유기 아지드와 말단 알키네스 사이의 1,3극성 사이클로어드 더하기(즉, Huisgen 사이클로어드 더하기)는 바이오 통합에 널리 이용되어 왔다.
구리 촉매
일반적으로 후이젠 반응은 가벼운 상태에서는 쉽게 진행되지 않는다. 멜달 외 연구진 및 샤플리스 외 연구진은 생리적 조건(중립 pH, 상온 및 물 용액)을 포함하여 매우 쉽게 경미하게 진행되는 Huisgen 반응의 구리(I) 분석 버전 CuAAC(Copper-catalized Azide-Alkyne Cycloadation의 경우)를 독립적으로 개발했다.[63][64] 이러한 반응은 생체 직교 반응도 있다. 아지드 및 알키네 모두 생물학적 시스템에서 일반적으로 존재하지 않기 때문에 이러한 기능성은 세포 환경에서도 화학적으로 반응할 수 있다. 그들은 또한 자연에서 발견되는 다른 기능 그룹과 반응하지 않기 때문에 생물 시스템을 동요시키지 않는다. 그 반응은 "클릭" 화학이라고 불릴 정도로 다재다능하다. 구리(I)는 독성이 있지만 세포독성을 감소시키고 CuAAC의 비율을 향상시키기 위해 많은 보호 리간드가 개발되어 체내 연구에 사용될 수 있게 되었다.[65]

예를 들어, 베르토지 외 연구진은 아지드화 기능화된 사카리드가 세포막의 글리칸에 대사 결합되고, 후속적으로 플루오포레알킨 결합으로 라벨을 붙이는 것을 보고했다. 현재 형광 라벨로 표시된 세포막은 현미경으로 이미징할 수 있다.[66]

변형률촉진 사이클로어다이드
구리(I)의 독성을 피하기 위해 베르토지 등은 유기 아지드와 변형된 사이클로크티네 사이의 변형률 촉진 아지드-알킨 사이클로어데이드(SPAAC)를 개발했다. 사이클로크티네의 각도 왜곡은 활성화 스트레인을 줄이고 상호작용을 강화하여 반응 속도를 높이는 데 도움을 주어 촉매 없이도 생리학적 조건에서 사용할 수 있다.[67]

예를 들어, 팅 등은 세포 표면의 특정 단백질에 리가아제 효소를 사용하여 아지도 기능을 도입했다. 그런 다음 아지드 태그가 붙은 단백질에는 사이클로크티네-플루오로포레 접합체가 붙어 형광 라벨이 붙은 단백질을 생산한다.[68]

참조
- ^ Huisgen, Rolf (1963). "1.3-Dipolare Cycloadditionen Ruckschau und Ausblick". Angewandte Chemie. 75 (13): 604–637. doi:10.1002/ange.19630751304.
- ^ a b Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions". Angewandte Chemie International Edition. 2 (11): 633–645. doi:10.1002/anie.196306331.
- ^ Firestone, R (1968). "Mechanism of 1,3-dipolar cycloadditions". Journal of Organic Chemistry. 33 (6): 2285–2290. doi:10.1021/jo01270a023.
- ^ Huisgen, Rolf (1976). "1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates". Journal of Organic Chemistry. 41 (3): 403–419. doi:10.1021/jo00865a001.
- ^ Mloston, G.; Langhals, E.; Huisgen, Rolf (1986). "First Two-Step 1,2-Dipolar Cycloadditons: Nonstereospecificity". J. Am. Chem. Soc. 108 (20): 6401–66402. doi:10.1021/ja00280a053.
- ^ Seyyed Amir, Siadati (2015). "An example of a stepwise mechanism for the catalyst-free 1,3-dipolar cycloaddition between a nitrile oxide and an electron rich alkene". Tetrahedron Letters. 56 (34): 4857–4863. doi:10.1016/j.tetlet.2015.06.048.
- ^ Huisgen, Rolf (1963). "1,3-Dipolar Cycloadditions. Past and Future". Angewandte Chemie International Edition. 2 (10): 565–598. doi:10.1002/anie.196305651.
- ^ Cox, A; Thomas, L; Sheridan, J (1958). "Microwave Spectra of Diazomethane and its Deutero Derivatives". Nature. 181 (4614): 1000–1001. Bibcode:1958Natur.181.1000C. doi:10.1038/1811000a0. S2CID 4245746.
- ^ Hilberty, P; Leforestier, C (1978). "Expansion of molecular orbital wave functions into valence bond wave functions. A simplified procedure". Journal of the American Chemical Society. 100 (7): 2012–2017. doi:10.1021/ja00475a007.
- ^ McGarrity, J.F.; Patai, Saul (1978). Basicity, acidity and hydrogen bonding. Diazonium and Diazo Groups. 1. pp. 179–230. doi:10.1002/9780470771549.ch6. ISBN 9780470771549.
- ^ Berner, Daniel; McGarrity, John (1979). "Direct observation of the methyldiazonium ion in fluorosulfuric acid". Journal of the American Chemical Society. 101 (11): 3135–3136. doi:10.1021/ja00505a059.
- ^ Muller, Eugen; Rundel, Wolfgans (1956). "Untersuchungen an Diazomethanen, VI. Mitteil.: Umsetzung von Diazoäthan mit Methyllithium". Chemische Berichte. 89 (4): 1065–1071. doi:10.1002/cber.19560890436.
- ^ Geittner, Jochen; Huisgen, Rolph; Reissig, Hans-Ulrich (1978). "Solvent Dependence of Cycloaddition Rates of Phenyldiazomethane and Activation Parameters". Heterocycles. 11: 109–120. doi:10.3987/S(N)-1978-01-0109.
- ^ Huisgen, Rolph; Reissig, Hans-Ulrich; Huber, Helmut; Voss, Sabine (1979). "α-Diazocarbonyl compounds and enamines - a dichotomy of reaction paths". Tetrahedron Letters. 20 (32): 2987–2990. doi:10.1016/S0040-4039(00)70991-9.
- ^ Sustmann, R (1974). "Orbital energy control of cycloaddition reactivity". Pure and Applied Chemistry. 40 (4): 569–593. doi:10.1351/pac197440040569.
- ^ Geittner, Jochen; Huisgen, Rolf (1977). "Kinetics of 1,3-dipolar cycloaddition reactions of diazomethane; A correlation with homo-lumo energies". Tetrahedron Letters. 18 (10): 881–884. doi:10.1016/S0040-4039(01)92781-9.
- ^ Huisgen, Rolf; Szeimies, Gunter; Mobius, Leander (1967). "K1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen". Chemische Berichte. 100 (8): 2494–2507. doi:10.1002/cber.19671000806.
- ^ Williamson, D. G.; Cvetanovic, R. J. (1968). "Rates of ozone-olefin reactions in carbon tetrachloride solutions". Journal of the American Chemical Society. 90 (14): 3668–3672. doi:10.1021/ja01016a011.
- ^ Bihlmaier, Werner; Geittner, Jochen; Huisgen, Rolf; ReissigP, Hans-Ulrich (1978). "The Stereospecificity of Diazomethane Cycloadditions". Heterocycles. 10: 147–152. doi:10.3987/S-1978-01-0147.
- ^ Huisgen, Rolf; Scheer, Wolfgang; Huber, Helmut (1967). "Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides". Journal of the American Chemical Society. 89 (7): 1753–1755. doi:10.1021/ja00983a052.
- ^ Dahmen, Alexander; Hamberger, Helmut; Huisgen, Rolf; Markowski, Volker (1971). "Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides". Journal of the Chemical Society D: Chemical Communications (19): 1192–1194. doi:10.1039/C29710001192.
- ^ Padwa, Albert; Smolanoff, Joel (1971). "Photocycloaddition of arylazirenes with electron-deficient olefins". Journal of the American Chemical Society. 93 (2): 548–550. doi:10.1021/ja00731a056.
- ^ Iwashita, Takashi; Kusumi, Takenori; Kakisawa, Hiroshi (1979). "A Synthesis of dl-isoretronecanol". Chemistry Letters. 8 (11): 1337–1340. doi:10.1246/cl.1979.1337.
- ^ Wang, Chia-Lin; Ripka, William; Confalone, Pat (1984). "A short and stereospecific synthesis of (±)-α-lycorane". Tetrahedron Letters. 25 (41): 4613–4616. doi:10.1016/S0040-4039(01)91213-4.
- ^ Kanemasa, Shuji (2002). "Metal-Assisted Stereocontrol of 1,3-Dipolar Cycloaddition Reactions". Synlett. 2002 (9): 1371–1387. doi:10.1055/s-2002-33506.
- ^ Bode, Jeffrey; Carreira, Erick (2011). "Stereoselective Syntheses of Epothilones A and B via Directed Nitrile Oxide Cycloaddition". Journal of the American Chemical Society. 123 (15): 3611–3612. doi:10.1021/ja0155635. PMID 11472140.
- ^ Vsevolod V. Rostovtsev; Luke G. Green; Valery V. Fokin; K. Barry Sharpless (2002). "A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes". Angewandte Chemie International Edition. 41 (14): 2596–22599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546.
- ^ Caramella, Pierluigi; Houk, K.N. (1976). "Geometries of nitrilium betaines. The clarification of apparently anomalous reactions of 1,3-dipoles". Journal of the American Chemical Society. 98 (20): 6397–6399. doi:10.1021/ja00436a062.
- ^ Caramella, Pierluigi; Gandour, Ruth W.; Hall, Janet A.; Deville, Cynthia G.; Houk, K. N. (1977). "A derivation of the shapes and energies of the molecular orbitals of 1,3-dipoles. Geometry optimizations of these species by MINDO/2 and MINDO/3". Journal of the American Chemical Society. 99 (2): 385–392. doi:10.1021/ja00444a013.
- ^ Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions". Angewandte Chemie International Edition. 2 (11): 633–645. doi:10.1002/anie.196306331.
- ^ Padwa, Albert (1983). 1,3-Dipolar Cycloaddition Chemistry. General Heterocyclic Chemistry Series. 1. United States of America: Wiley-Interscience. pp. 141–145. ISBN 978-0-471-08364-1.
- ^ Koszinowski, J. (1980). thesis (PhD Thesis).
- ^ Evans, David; Ripin, David; Halstead, David; Campos, Kevin (1999). "Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide". Journal of the American Chemical Society. 121 (29): 6816–6826. doi:10.1021/ja990789h.
- ^ M=C 및 M=N 본드의 합성반응: Ylide 형성, 재배열 및 1,3-Dipolar Cycloaddition. 히야마, T. W. J. 에드; 엘스비에, 2007; 제11권.
- ^ Padwa, Albert.; Hornbuckle, Susan F. (1991). "Ylide formation from the reaction of carbenes and carbenoids with heteroatom lone pairs". Chemical Reviews. 91 (3): 263–309. doi:10.1021/cr00003a001.
- ^ a b Janulis, Eugene P.; Arduengo, Anthony J. (1983). "Structure of an electronically stabilized carbonyl ylide". Journal of the American Chemical Society. 105 (18): 5929–5930. doi:10.1021/ja00356a044.
- ^ 프라카시, G. K. S.; 엘리스, R. W.; 펠버그, J. D.; 올라, G. A. 포름알데히드 0-메틸라이드, [CH2=O+-CH2: 부모 카보닐 일라이드] J 암 화학 Soc 1986, 108, 1341.
- ^ Sammes, P. G.; Street, L. J. Oxido Pyrylium Ylides J와 함께 분자 내 사이클로 첨가. Chem. Soc, Chem. 코뮌 1982년 1056년
- ^ 가스트, M. E.; 맥브라이드, B. J.; 2-(Ω-알케닐)-5-하이드록시-4-피이드로네 테트라헤드론 레트. 1983, 24, 1675.
- ^ Gisch, John F.; Landgrebe, John A. (1985). "Dichlorocarbene from flash vacuum pyrolysis of trimethyl(trichloromethyl)silane. Possible observation of 1,1-dichloro-3-phenylcarbonyl ylide". The Journal of Organic Chemistry. 50 (12): 2050–2054. doi:10.1021/jo00212a009.
- ^ Huan, Zhenwei; Landgrebe, John A.; Peterson, Kimberly (1983). "Dibromocarbonyl ylides. Deoxygenation of aldehydes and ketones by dibromocarbene". The Journal of Organic Chemistry. 48 (24): 4519–4523. doi:10.1021/jo00172a015.
- ^ Martin, Charles W.; Lund, Paul R.; Rapp, Erich; Landgrebe, John A. (1978). "Halogenated carbonyl ylides in the reactions of mercurial dihalocarbene precursors with substituted benzaldehydes". The Journal of Organic Chemistry. 43 (6): 1071–1076. doi:10.1021/jo00400a009.
- ^ Hodgson, D. M.; Bruckl, T.; Glen, R.; Labande, A. H.; Selden, D. A.; Dossetter, A. G.; Redgrave, A. J. Catalytic enantioselective intermolecular cycloadditions of 2-diazo-3,6-diketoester-derived carbonyl ylides with alkene dipolarophiles Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 5450.
- ^ Padwa, Albert; Hertzog, Donald L.; Nadler, William R. (1994). "Intramolecular Cycloaddition of Isomunchnone Dipoles to Heteroaromatic .pi.-Systems". The Journal of Organic Chemistry. 59 (23): 7072–7084. doi:10.1021/jo00102a037.
- ^ 하마구치, M.; 이바타, T. 새로운 형태의 메소이온 시스템. 에틸렌 화합물인 이소문첸의 1,3-다극성 사이클로어 추가 1975, 499.
- ^ Park, Soon-Bong; Sakata, Naoya; Nishiyama, Hisao (1996). "Aryloxycarbonylcarbene Complexes of Bis(oxazolinyl)pyridineruthenium as Active Intermediates in Asymmetric Catalytic Cyclopropanations". Chemistry - A European Journal. 2 (3): 303–306. doi:10.1002/chem.19960020311.
- ^ Snyder, James P.; Padwa, Albert; Stengel, Thomas; Arduengo, Anthony J.; Jockisch, Alexander; Kim, Hyo-Joong (2001). "A Stable Dirhodium Tetracarboxylate Carbenoid: Crystal Structure, Bonding Analysis, and Catalysis". Journal of the American Chemical Society. 123 (45): 11318–11319. doi:10.1021/ja016928o. PMID 11697986.
- ^ a b Hodgson, D. M.; Pierard, F. Y. T. M.; Stupl, P. A. 촉매변환성항체재배열재배열재배열재배열 및 디아조 화합물 Chem Soc 2001, 30, 50.
- ^ Yoshikai, Naohiko; Nakamura, Eiichi (2003). "Theoretical Studies on Diastereo- and Enantioselective Rhodium-Catalyzed Cyclization of Diazo Compoundvia Intramolecular C—H Bond Insertion". Advanced Synthesis & Catalysis. 345 (910): 1159–1171. doi:10.1002/adsc.200303092.
- ^ Nakamura, Eiichi; Yoshikai, Naohiko; Yamanaka, Masahiro (2002). "Mechanism of C−H Bond Activation/C−C Bond Formation Reaction between Diazo Compound and Alkane Catalyzed by Dirhodium Tetracarboxylate". Journal of the American Chemical Society. 124 (24): 7181–7192. doi:10.1021/ja017823o. PMID 12059244.
- ^ 코스탄티노, G.; 로비토, R.; 맥키아룰로, A.; 펠리치아리, R. 디르호듐(Dirhodium)에서 파생된 금속-카르브노이드 중간자 구조(R. Structure of metal-carbinoider)II) α-diazocarbonyl 화합물의 테트라카르복실산염 매개 분해: DFT 연구 J Mol Struc-Thechomic 2002, 581, 111.
- ^ a b c d M. Hodgson, D.; H. Labande, A.; Muthusamy, S. 유기농 반응에서; John Wiley & Sons, Inc.: 2004.
- ^ Suga, Hiroyuki; Ebiura, Yasutaka; Fukushima, Kazuaki; Kakehi, Akikazu; Baba, Toshihide (2005). "Efficient Catalytic Effects of Lewis Acids in the 1,3-Dipolar Cycloaddition Reactions of Carbonyl Ylides with Imines". The Journal of Organic Chemistry. 70 (26): 10782–10791. doi:10.1021/jo051743b. PMID 16356001.
- ^ a b Padwa, Albert; Fryxell, Glen E.; Zhi, Lin (1990). "Tandem cyclization-cycloaddition reaction of rhodium carbenoids. Scope and mechanistic details of the process". Journal of the American Chemical Society. 112 (8): 3100–3109. doi:10.1021/ja00164a034.
- ^ Houk, K. N.; Sims, Joyner.; Duke, R. E.; Strozier, R. W.; George, John K. (1973). "Frontier molecular orbitals of 1,3 dipoles and dipolarophiles". Journal of the American Chemical Society. 95 (22): 7287–7301. doi:10.1021/ja00803a017.
- ^ Houk, K. N.; Rondan, Nelson G.; Santiago, Cielo; Gallo, Catherine J.; Gandour, Ruth Wells; Griffin, Gary W. (1980). "Theoretical studies of the structures and reactions of substituted carbonyl ylides". Journal of the American Chemical Society. 102 (5): 1504–1512. doi:10.1021/ja00525a006.
- ^ Padwa, Albert; Weingarten, M. David (1996). "Cascade Processes of Metallo Carbenoids". Chemical Reviews. 96 (1): 223–270. doi:10.1021/cr950022h. PMID 11848752.
- ^ Padwa, Albert; Austin, David J. (1996). "Ligand-Induced Selectivity in the Rhodium(II)-Catalyzed Reactions of α-Diazo Carbonyl Compounds†". The Journal of Organic Chemistry. 61: 63–72. doi:10.1021/jo951576n.
- ^ 슈가, H.; 카케히, A.; Ito, S.; Inoue, K.; Ishida, H.; Ibata, T. Stereocontrol in a Ytterbium Triflate-Catalyzed 1,3-Dipolar Cycloaddition Reaction of Carbonyl Ylide with N-Substituted Maleimides and Dimethyl Fumarate B Chem Soc Jpn 2001, 74, 1115.
- ^ Geng, Zhe; Chen, Bin; Chiu, Pauline (2006). "Total Synthesis of Pseudolaric Acid A". Angewandte Chemie International Edition. 45 (37): 6197–6201. doi:10.1002/anie.200602056. PMID 16906616.
- ^ Suga, Hiroyuki; Inoue, Kei; Inoue, Shuichi; Kakehi, Akikazu; Shiro, Motoo (2005). "Chiral 2,6-Bis(oxazolinyl)pyridine−Rare Earth Metal Complexes as Catalysts for Highly Enantioselective 1,3-Dipolar Cycloaddition Reactions of 2-Benzopyrylium-4-olates". The Journal of Organic Chemistry. 70 (1): 47–56. doi:10.1021/jo049007f. PMID 15624905.
- ^ Onishi, Tomoyuki; Sebahar, Paul; Williams, Robert (2003). "Concise, Asymmetric Total Synthesis of Spirotryprostatin A". Organic Letters. 5 (17): 3135–3137. doi:10.1021/ol0351910. PMID 12917000.
- ^ Tornoe, Christian; Christensen, Caspar; Meldal, Morten (2002). "Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides". Journal of Organic Chemistry. 67 (9): 3057–3064. doi:10.1021/jo011148j. PMID 11975567.
- ^ Rostovtsev, Vsevolod; Green, Luke; Fokin, Valery; Sharpless, Barry K. (2002). "A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes". Angewandte Chemie International Edition. 41 (14): 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546.
- ^ Besanceney-Webler, Christen; Jiang, Hao; Zheng, Tianqing; Feng, Lei; Soriano del Amo, David; Wang, Wei; Klivansky, Liana M.; Marlow, Florence L.; Liu, Yi; Wu, Peng (2011). "Increasing the Efficacy of Bioorthogonal Click Reactions for Bioconjugation: A Comparative Study". Angewandte Chemie International Edition. 50 (35): 8051–8056. doi:10.1002/anie.201101817. PMC 3465470. PMID 21761519.
- ^ Breidenbach, Mark; Gallagher, Jennifer; King, David; Smart, Brian; Wu, Peng; Bertozzi, Carolyn (2010). "Targeted metabolic labeling of yeast N-glycans with unnatural sugars". Proceedings of the National Academy of Sciences of the United States of America. 107 (9): 3988–3993. Bibcode:2010PNAS..107.3988B. doi:10.1073/pnas.0911247107. PMC 2840165. PMID 20142501.
- ^ Agard, Nicholas; Prescher, Jennifer; Bertozzi, Carolyn (2004). "A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems". Journal of the American Chemical Society. 126 (46): 15046–15047. doi:10.1021/ja044996f. PMID 15547999.
- ^ Fernandez-Suarez, Marta; Baruah, Hemanta; Martinez-Hernandez, Laura; Xie, Kathleen; Baskin, Jeremy; Bertozzi, Carolyn; Ting, Alice (2007). "Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes". Nature Biotechnology. 25 (12): 1483–1487. doi:10.1038/nbt1355. PMC 2654346. PMID 18059260.


