양이온-피 상호작용
Cation–pi interaction
양이온-수분자 상호작용은 전자가 풍부한 π 시스템(예: 벤젠, 에틸렌, 아세틸렌)의 면과 인접한 양이온(예+: Li+, Na) 사이의 비균형 분자 상호작용이다. 이 상호작용은 단극(cation)과 사극(supdedpole) 사이의 비균질 결합의 한 예다. 본딩 에너지는 용액 위상 값이 수소 본딩 및 염교와 동일한 크기 순서에 속하므로 유의하다. 이러한 다른 비균등 결합과 유사하게, 양이온-효소 상호작용은 특히 단백질 구조, 분자 인식 및 효소 촉매변환에서 중요한 역할을 한다. 그 효과는 또한 관찰되어 합성 시스템에도 사용되었다.[1][2]

효과의 기원
모델 π 시스템인 벤젠은 분자 대칭으로 인해 약하게 극지방 탄소-수소 결합의 기여가 취소되기 때문에 영구적인 쌍극자 모멘트가 없다. 그러나 벤젠 링 위와 아래의 전자가 풍부한 π 시스템은 부분 음전하를 발생시킨다. 역밸런싱 양전하가 벤젠 원자의 평면과 연관되어 전기적 4중극(쌍쌍쌍의 쌍극, 평행사변형처럼 정렬되어 순분자 쌍극 모멘트가 없다. 4극의 음전하 부위는 양전하 종과 우호적으로 상호작용할 수 있다. 특히 높은 전하 밀도의 양이온에서 강한 효과가 관찰된다.[2]
양이온-수분 상호작용의 특성
가장 많이 연구된 양이온-산화 상호작용은 방향족 π 시스템과 알칼리 금속 또는 질소 양이온 사이의 결합을 포함한다. 최적의 상호작용 기하학적 구조는 6배 축을 따라 π면 상단을 중심으로 반데르 발스(Van der Waals)가 방향족 링과 접촉하도록 배치한다.[3] 연구에 따르면 단순한 시스템에서의 상호작용을 전기공학이 지배하고, 상대 결합 에너지는 정전기 전위 에너지와 잘 상관관계가 있다.[4][5]
더거티와 동료들이 개발한 정전기 모델은 정전기 흡인력의 차이에 근거한 결합 에너지의 추세를 설명한다. cation-synchrones 쌍의 상호작용 에너지는 π 아레네 면 위의 정전기 전위와 잘 상관관계가 있는 것으로 밝혀졌다. 11 Na-aromatic+ adducts의 경우, 서로 다른 유도들 사이의 결합 에너지 변화는 정전기차에 의해 완전히 합리화될 수 있다. 실제로, 이것은 일련의 아레네에 대한 정전기적 전위 지도의 시각적 표현에 기초하여 질적으로 추세를 예측할 수 있게 한다. 정전기 흡인력은 양이온-배기 본딩의 유일한 구성요소가 아니다. 예를 들어, 1,3,5-트리플루오로벤젠은 사소한 쿼드폴 모멘트를 가지고 있음에도 불구하고 양이온과 상호작용을 한다. 비정전기력이 존재하는 동안, 이러한 구성 요소는 광범위한 영역에서도 유사한 상태를 유지하며, 정전기 모델은 상대 결합 에너지를 예측하는 데 유용한 도구가 된다. 구속력에 기여하는 다른 "효과"는 잘 이해되지 않는다. 양극화, 기증자-수용자[permanent dead link] 및 충전-이전 상호작용은 관련되었지만, 에너지적인 경향은 이러한 효과를 이용하는 영역과 양이온의 능력과는 잘 맞지 않는다. 예를 들어 유도 쌍극체가 제어 효과라면 사이클로헥산 같은 알리파틱 화합물은 좋은 양이온-염기 파트너(그러나 그렇지 않다)[4]가 되어야 한다.
양이온-변환 상호작용은 비균등성이며 따라서 전환 금속과 systems 시스템 사이의 결합과는 근본적으로 다르다. 전이 금속은 d-orbitals를 통해 π-systems와 전자 밀도를 공유할 수 있는 능력을 가지고 있어, 성격상 공밸런스(calent)가 높으며 cation-proble interaction으로 모델링할 수 없는 결합을 형성한다.
계간 결합 강도에 영향을 미치는 요인
몇 가지 기준은 결합의 강도에 영향을 미친다: 양이온의 특성, 용해 효과, π 시스템의 특성, 상호작용의 기하학.
양이온의 성질
전기 공학(Coulomb's law)에서, 더 작고 더 양전하된 양이온들은 더 큰 정전기 흡인력으로 이어진다. cation-properties 상호작용은 전기 공학에 의해 예측되므로, 전하 밀도가 큰 cation은 ations 시스템과 더 강하게 상호작용한다.
다음 표는 벤젠과 가스 단계의 여러 양이온 사이에 결합하는 일련의 깁스 자유 에너지를 보여준다.[2][6] 단일 충전 종의 경우 가스 위상 상호작용 에너지는 이온 반지름, r n 비구형 이온 반지름은 근사치임).[7][8]

M+ 리+ 나+ K+ NH4+ Rb+ NMe4+ –ΔG [kcal/mol] 38 27 19 19 16 9 [ [] 0.76 1.02 1.38 1.43 1.52 2.45
이 경향은 쿨롱 힘이 상호작용 강도에 중심적 역할을 한다는 생각을 지지한다. 왜냐하면 다른 유형의 결합의 경우 더 크고 더 편광 가능한 이온이 더 큰 결합 에너지를 가질 것으로 예상하기 때문이다.
용해 효과
용제의 성질은 또한 본딩의 절대 강도와 상대 강도를 결정한다. 양이온-수분 상호작용에 대한 대부분의 데이터는 가스 단계에서 획득된다. 그 경우 유인이 가장 뚜렷하기 때문이다. 간극-간극 상호작용에 의해 얻어진 에너지는 용해 에너지 손실에 의해 부분적으로 상쇄되기 때문에 중간 용매 분자는 효과를 약화시킬 것이다.
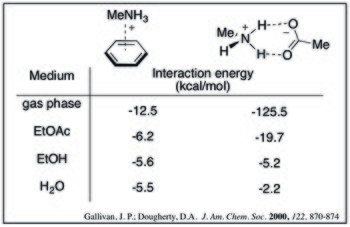
주어진 양이온-배열 인덕트의 경우, 상호작용 에너지는 용제 극성이 증가함에 따라 감소한다. 이는 다양한 용매에서 메틸람모늄과 벤젠의 다음과 같은 계산된 상호작용 에너지로 알 수 있다.[9]

또한 용도와 양이온 효과 사이의 절충은 일련의 양이온에 대한 교호작용 강도의 순서를 재정렬하게 한다. 가스 단계에서 가장 조밀하게 충전된 양이온이 가장 강력한 양이온-수분 상호작용을 하는 반면, 이온들은 또한 높은 황량화 벌칙을 가진다. 이는 알칼리 금속의 물에서 상대 양이온-배당 결합 강도에 의해 입증된다.[10]

π 제도의 특성
쿼드폴 모멘트
서로 다른 영역의 4극 모멘트를 비교하는 것은 교호작용 강도와 대략 상관관계가 있기 때문에 cation-transaction binding의 추세를 예측하는 데 유용한 정성적 도구다. 4중극 모멘트가 큰 아렌들은 일반적으로 결합에 더 능숙하다.
그러나 4극-이온 모델 시스템은 양이온-이온 교호작용을 정량적으로 모형화하는 데 사용할 수 없다. 그러한 모델은 점 전하를 가정하며, 따라서 단시간 간 결합 거리를 고려할 때 유효하지 않다. 에너지를 예측하기 위해 전기학을 사용하기 위해서는 단지 4극 모멘트가 아닌 정전기 전위 표면을 고려해야 한다.[2]
방향족 링의 대체품

대체물의 전자적 특성도 인력의 강도에 영향을 미친다.[11] 전자 인출 그룹(예: cyano -CN)은 상호작용을 약화시키는 반면, 전자 기부 대체물(예: 아미노 -NH2)은 양이온-염기 결합을 강화한다. 이 관계는 여러 대체자를 위한 여백에 정량적으로 설명되어 있다.
양이온-변환 결합 에너지의 전자적 경향은 아릴 반응성의 경향과 그다지 유사하지 않다. 실제로 대체물질의 공명 참여 효과는 아레네와의 많은 화학 반응에서 매우 중요함에도 불구하고 양이온-염색 결합에 실질적으로 기여하지 않는다. 이는 다양한 대체 아레네에 대한 cation-multi 상호작용 강도가 σmeta Hammett 매개변수와 상관관계가 있다는 관측에 의해 입증되었다. 이 매개변수는 아릴 링에 작용하는 기능 그룹의 유도 효과를 설명하기 위한 것이다.[4]
양이온-배당 상호작용에서 대체효과의 기원은 종종 전자 기증이나 π 시스템 내부 또는 외부로부터의 인출에 의한 양극화에 기인한다.[12] 이 설명은 직관적으로 이해가 되지만, 이후의 연구는 결함이 있다는 것을 보여주었다. 휠러와 후크의 최근 계산 연구는 그 영향이 주로 양이온과 대체 쌍극자 사이의 공간 간 직접 상호작용에 기인한다는 것을 강하게 보여준다. 본 연구에서는 대체물이 (여분의 수소 원자에 대해 보정)될 위치의 "H-X" 분자와의 상호작용과 미신분 벤젠을 모델링한 계산이 거의 모든 양이온-증가 결합 추세를 차지했다. 매우 강한 파이 기증자나 수용자의 경우, 이 모델은 전체 상호작용을 설명하지 못했다. 이러한 경우 양극화가 더 중요한 요소가 될 수 있다.[5]
이형 자동 시스템으로 바인딩
헤테로사이클은 종종 헤테로아톰의 외로운 쌍이 방향계(예: 인도어, 피롤)에 통합될 때 양이온 결합을 향해 활성화된다. 반대로, 외로운 쌍이 방향성에 기여하지 않을 때(예: 피리딘) 이형 원자의 전기성이 이기고 양이온-복제 결합 능력을 약화시킨다.
고전적으로 "전자가 풍부한" 몇몇 이질체들은 양이온-배당 결합에 관한 한 가난한 기증자들이기 때문에, 이질체 반응성 추세를 바탕으로 한 양이온-배당 추세를 예측할 수 없다. 다행히도, 전술한 미묘함은 관련 이질체의 정전기적 전위 표면에서 나타난다.[2]

cation-csocycle 상호작용은 항상 cation-synchronization 상호작용은 아니다. 어떤 경우에는 이온이 한 쌍에 직접 결합되는 것이 더 유리하다. 예를 들어, 피리딘-나+ 콤플렉스에서는 이런 경우가 있다고 생각된다.
기하학
cation-properties 교호작용은 대략 거리 의존도가 1/r이다n(n<2). 상호작용은 1/r3 의존성을 갖는 단순한 이온-콰드루폴 상호작용보다 거리에 덜 민감하다.[13]
세릴과 동료들의 연구는 양이온이 원자 평면에 수직으로 위치할 때 양이온-수동 교호작용이 가장 강하다는 것을 확인하면서 상호작용의 기하학을 더 자세히 조사했다. 이 기하학과의 변화는 여전히 significant 각도가 90도에 근접함에 따라 약해지는 유의한 상호작용을 나타낸다. 오프 축 상호작용의 경우 선호되는 ϕ은 두 H 원자 사이에 cation을 배치한다. 평형 본드 거리 또한 오프 축 각도에 따라 증가한다. 양이온과 탄소 링의 일렬로 있는 에너지는 잠재적 에너지 표면의 안장 지점이며, 이는 양이온과 4극의 양의 영역 사이의 상호작용이 이상적이지 않다는 생각과 일치한다.[14]

상대 상호작용 강도
수용성 매체에서 양이온-수분 상호작용은 암모늄-카르복실산염 교량(잠재적으로 강한)과 비교된다. 아래 계산된 값은 용제 극성이 증가함에 따라 양이온-배열 복합체의 강도가 극적으로 감소한다는 것을 보여준다. 이러한 경향은 황폐화 효과에 의해 합리화될 수 있다: 소금 교량 형성은 두 충전된 종에 대해 높은 황폐화 벌점을 가지고 있는 반면, 양이온-염화 복합체는 양이온에 대해 상당한 벌점만 지불할 것이다.[9]
자연에서
자연의 빌딩 블록에는 향기로운 모이티가 풍부하게 들어 있다. 최근, 한때 자연에서 순수 소수성이라고 생각되었던 많은 구조적인 특징들이 사실 cation-the consistance interaction에 관여하고 있다는 것이 명백해졌다. 페닐알라닌, 트립토판, 티로신, 히스티딘의 아미노산 사이드 체인은 충전된 아미노산 사이드 체인, 금속 이온, 소분자 신경전달물질, 제약제 등과 같은 양이온 종에 결합할 수 있다. 실제로 음이온 집단을 포함하도록 가설을 세운 고분자 결합 부위(양식에 대한 친화력을 바탕으로 함)는 여러 경우에서 방향족 잔류물로 구성된 것으로 밝혀졌다. 양이온-염기 상호작용은 질소성 측면 체인의 pKa를 조절하여 양성자 형태의 풍부함을 증가시킬 수 있다. 이는 단백질 구조와 기능에 영향을 미친다.[15] 이러한 맥락에서 연구된 것은 적지만, DNA 염기 또한 양이온-염색 상호작용에 참여할 수 있다.[16][17]
단백질 구조에서의 역할
양이온과 양이온 상호작용이 단백질 구조에 영향을 미친다는 초기 증거는 결정학적 데이터에서 방향족 사이드 체인이 불균형 빈도로 질소가 함유된 사이드 체인(양성, 양이온 종으로 존재할 수 있음)과 밀접하게 접촉하여 나타난다는 관찰이었다.
1986년 벌리와 펫스코가 발표한 연구에서 다양한 단백질 세트를 살펴본 결과 방향족 잔류물인 Phe, Tyr, Trp의 약 50%가 아미노군 6å 이내였다. 또한, 사이드 체인 라이스, 아스앤, 글렌 및 히스가 함유된 질소의 약 25%는 밴 데르 발스 내에 있었고, 아그의 50%는 여러 방향족 잔류물과 접촉했다(평균 2개).[18]
더 큰 데이터 세트에 대한 연구 결과, 양식과 방향족 사이드 체인의 극적인 배열 등 유사한 추세가 나타났다. N-H 수력발전소를 방향족 잔류물 쪽으로 정렬하는 경우도 있었고, 계통계 위에 계통 계량제를 쌓는 경우도 있었다. 특히 아르그와 trp의 긴밀한 접촉에 대해 강한 경향이 발견되었다. 특히 아르그(Arg)의 구아니디늄 모이티(Guanidinium moiety)는 방향족 잔류물 위에 쌓이는 성향이 높은 동시에 인근 산소원자와 수소결합도 한다.[19][20][21]
분자 인식 및 신호

분자 인식에서 양이온-염기 상호작용의 예는 양이온-염기 암모늄에 대한 내생 리간드, 아세틸콜린(양전하 분자)을 결합하는 니코틴 아세틸콜린 수용체(nAChR)에서 볼 수 있다. nACHR 신경수용기는 아세틸콜린 결합 시 개방되는 잘 연구된 리간드 게이트 이온 채널이다. 아세틸콜린 수용체는 파킨슨병, 알츠하이머병, 정신분열증, 우울증, 자폐증 등 신경학적 질환이 많이 발생하는 치료 대상이다. 더거티와 동료들의 연구는 키 트립토판 잔류물에 특정한 구조적 변화를 만들고 활성도 결과를 계간-변환 결합 능력과 상관시킴으로써 계간-변환 상호작용이 nACHR을 결합하고 활성화하는 데 중요하다는 것을 확인했다.[22]
nACHR은 특히 뇌에서 니코틴을 결합시키는 데 중요하며, 니코틴 중독에 핵심적인 역할을 한다. 니코틴은 특히 양성자일 때 아세틸콜린과 유사한 약리성을 가지고 있다. 강력한 증거는 니코틴이 근육 활동에 영향을 주지 않고 뇌 수용체를 선택적으로 활성화할 수 있는 능력의 중심이 되는 양이온-증상 상호작용을 뒷받침한다.[23][24]

또 다른 예는 UV-B 감지 단백질 UVR8 식물에서 볼 수 있다. 여러 트립토판 잔류물은 아르기닌 잔류물과 양이온-증식 상호작용을 통해 상호작용하며, 아르기닌 잔류물은 다시 염교에서 단백질의 두 번째 복사본에 산성 잔류물을 함유한다. 트립토판 잔류물에 의한 광자 흡수가 이러한 상호작용을 방해하고 단백질 조광기의 분리를 유도하는 것이 제안되었다[25].
Cation-tency 바인딩은 세포 표면 인식에서도[2][26] 중요한 것으로 간주된다.
효소 촉매제
양이온-증가 상호작용은 전이 상태에서 양의 전하 축적을 안정화함으로써 화학 반응을 촉진할 수 있다. 이러한 종류의 효과는 효소 시스템에서 관찰된다. 예를 들어 아세틸콜린 에스테라아제는 활성 부위에서 분기 암모늄을 결합하는 중요한 방향족 그룹을 포함한다.[2]
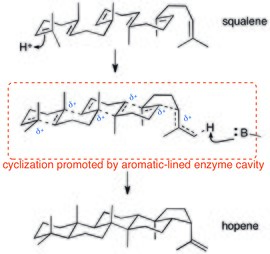
폴리사이클로화 효소 또한 양이온-염기 상호작용에 의존한다. 스칼렌의 양성자 트리거 폴리실라이제이션은 (잠재적으로 결합되는) 양이온 캐스케이드를 통해 진행되기 때문에 양이온-수분 상호작용은 분산된 양전하를 안정화하는 데 이상적이다. 스칼린-호펜 사이클라아제의 결정 구조는 활성 부위가 방향제 잔류물로 줄지어 있음을 보여준다.[27]
합성 시스템에서
솔리드 스테이트 구조
양이온-염기 상호작용은 합성 분자의 결정에서도 관찰되었다. 예를 들어 아오키와 동료들은 인도레-3-아세트산 콜린 에스테르와 충전되지 않은 아날로그의 고체 구조를 비교했다. 충전된 종에서, 격자 내 인접 분자의 교호작용뿐만 아니라, 교호작용과 교호작용도 관찰된다. 이소스테릭 중성 화합물의 결정에서는 동일한 접힘이 관찰되지 않으며, 테르트 부틸 그룹과 인접한 인들 사이에는 어떠한 상호 작용도 없다.[28]

초분자 수용체
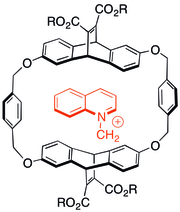
계간-간 상호작용에 대한 첫 번째 연구들 중 일부는 사이클로판 숙주-게스트 화학에서 질소성 충전 분자의 상호작용을 조사하는 것을 포함했다. 방향족 숙주 캡슐에 음이온성 용해성 그룹을 첨가했을 때도 양이온성 손님들은 π-system과의 연계를 선호하는 경우가 많은 것으로 나타났다. 오른쪽에 보이는 호스트 유형도 N-알킬화 반응을 촉진시켜 양이온 제품을 형성할 수 있었다.[29]
보다 최근, 계간 중심 기질 결합과 강직화는 레이먼드와 버그먼이 개발한 초분자 금속 리간드 클러스터 촉매 시스템에 관련되어 있다.[30]
초분자 조립체에서 π-π, CH-π 및 ation-cation 상호작용 사용
π-systems은 다양한 기능 그룹과의 다용도 비균형 상호작용 때문에 초분자 조립체에서 중요한 구성 요소다. 특히 cular-π, CH-π, π-cation 상호작용은 초분자 조립과 인식에 널리 사용된다.

π-π 상호작용은 두 &pi-interaction과 cation-interaction 사이의 직접적인 상호작용과 관련이 있다. cation-interaction은 π-system 면과의 정전기적 상호작용에서 발생한다. 이 두 상호작용과는 달리 CH-변환 상호작용은 주로 C-H 궤도와 and계 사이의 전하 전달에서 발생한다.
초분자 어셈블리에 π-π 교호작용을 적용하는 주목할 만한 예는 카테난의 합성이다. 카테난의 합성을 위한 주요 과제는 분자를 통제된 방식으로 연동시키는 것이다. 스토다트와 동료들은 전자가 풍부한 벤젠 유도체와 전자가 부족한 피리디늄 고리 사이의 강력한 π-π 상호작용을 활용한 일련의 시스템을 개발했다.[31] [2]카타넨은 bis(피리디늄) (A), bisparaphenilen-34-crown-10 (B), 1, 4-bis(브로메틸)벤젠 C (그림 2)를 반응시켜 합성되었다. A와 B의 π-π 상호작용은 [2]카테난 제품을 생성하기 위해 복합 C와의 대체 반응에 의해 추가로 사이클링된 연동형 템플릿 중간을 형성하도록 지시하였다.

유기합성 및 촉매제
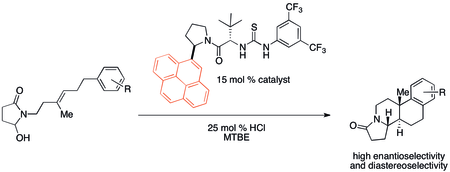
cation-properties 상호작용은 역사적으로 수많은 유기적 반응에서 눈에 띄지 않지만 중요했을 가능성이 높다. 그러나 최근 촉매 설계의 잠재적 적용에 관심이 모아지고 있다. 특히, 비동축 유기 촉매들은 cation-properties 바인딩 특성과 상관관계가 있는 반응성 및 선택성 추세를 보이는 것으로 밝혀졌다. Jacobsen과 동료들이 개발한 폴리사이클링은 아래에 표시된 촉매를 사용하여 특히 강한 양이온-증가 효과를 나타낸다.[32]
음이온과 음이온의 상호작용

많은 측면에서 음이온과 음이온의 상호작용은 기본 원칙은 동일하지만 양이온-음향 상호작용과는 반대다. 현재까지 알려진 예는 상당히 적다. 음전하를 끌어들이기 위해서는 π계통의 전하분포를 되돌려야 한다. 이것은 π계(예: 헥사플루오로벤젠)를 따라 몇 개의 강한 전자 인출 대체물을 배치함으로써 달성된다.[33] 음이온-효과는 특정 음이온에 대한 화학 센서에서 유리하게 이용된다.[34]
참고 항목
참조
- ^ Eric V. Anslyn; Dennis A. Dougherty (2004). Modern Physical Organic Chemistry. University Science Books. ISBN 978-1-891389-31-3.
- ^ a b c d e f g Dougherty, D. A.; J.C. Ma (1997). "The Cation–π Interaction". Chemical Reviews. 97 (5): 1303–1324. doi:10.1021/cr9603744. PMID 11851453.
- ^ Tsuzuki, Seiji; Yoshida, Masaru; Uchimaru, Tadafumi; Mikami, Masuhiro (2001). "The Origin of the Cation/π Interaction: The Significant Importance of the Induction in Li+and Na+Complexes". The Journal of Physical Chemistry A. 105 (4): 769–773. Bibcode:2001JPCA..105..769T. doi:10.1021/jp003287v.
- ^ a b c d S. Mecozzi; A. P. West; D. A. Dougherty (1996). "Cation–π Interactions in Simple Aromatics: Electrostatics Provide a Predictive Tool". JACS. 118 (9): 2307–2308. doi:10.1021/ja9539608.
- ^ a b S. E. Wheeler; K. N. Houk (2009). "Substituent Effects in Cation/π Interactions and Electrostatic Potentials above the Center of Substituted Benzenes Are Due Primarily to through-Space Effects of the Substituents". J. Am. Chem. Soc. 131 (9): 3126–7. doi:10.1021/ja809097r. PMC 2787874. PMID 19219986.
- ^ J. C. Amicangelo; P. B. Armentrout (2000). "Absolute Binding Energies of Alkali-Metal Cation Complexes with Benzene Determined by Threshold Collision-Induced Dissociation Experiments and ab Initio Theory". J. Phys. Chem. A. 104 (48): 11420–11432. Bibcode:2000JPCA..10411420A. doi:10.1021/jp002652f.
- ^ 로빈슨 RA, 스토크스 RH 전해액. 영국: 1959년 피트만, 버터워스 출판사.
- ^ Clays and Clay Minerals, Vol.45, No. 6 859-866, 1997.
- ^ a b Gallivan, Justin P.; Dougherty, Dennis A. (2000). "A Computational Study of Cation−π Interactions vs Salt Bridges in Aqueous Media: Implications for Protein Engineering" (PDF). Journal of the American Chemical Society. 122 (5): 870–874. doi:10.1021/ja991755c.
- ^ Kumpf, R.; Dougherty, D. (1993). "A mechanism for ion selectivity in potassium channels: Computational studies of cation–pi interactions". Science. 261 (5129): 1708–10. Bibcode:1993Sci...261.1708K. doi:10.1126/science.8378771. PMID 8378771.
- ^ Raju, Rajesh K.; Bloom, Jacob W. G.; An, Yi; Wheeler, Steven E. (2011). "Substituent Effects on Non-Covalent Interactions with Aromatic Rings: Insights from Computational Chemistry". ChemPhysChem. 12 (17): 3116–30. doi:10.1002/cphc.201100542. PMID 21928437.
- ^ Hunter, C. A.; Low, C. M. R.; Rotger, C.; Vinter, J. G.; Zonta, C. (2002). "Supramolecular Chemistry and Self-assembly Special Feature: Substituent effects on cation–pi interactions: A quantitative study". Proceedings of the National Academy of Sciences. 99 (8): 4873–4876. Bibcode:2002PNAS...99.4873H. doi:10.1073/pnas.072647899. PMC 122686. PMID 11959939.
- ^ Dougherty, Dennis A. (1996). "cation–pi interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp". Science. 271 (5246): 163–168. Bibcode:1996Sci...271..163D. doi:10.1126/science.271.5246.163. PMID 8539615. S2CID 9436105.
- ^ Marshall, Michael S.; Steele, Ryan P.; Thanthiriwatte, Kanchana S.; Sherrill, C. David (2009). "Potential Energy Curves for Cation−π Interactions: Off-Axis Configurations Are Also Attractive". The Journal of Physical Chemistry A. 113 (48): 13628–32. Bibcode:2009JPCA..11313628M. doi:10.1021/jp906086x. PMID 19886621.
- ^ Lund-Katz, S; Phillips, MC; Mishra, VK; Segrest, JP; Anantharamaiah, GM (1995). "Microenvironments of basic amino acids in amphipathic alpha-helices bound to phospholipid: 13C NMR studies using selectively labeled peptides". Biochemistry. 34 (28): 9219–9226. doi:10.1021/bi00028a035. PMID 7619823.
- ^ M. M. Gromiha; C. Santhosh; S. Ahmad (2004). "Structural analysis of cation–π interactions in DNA binding proteins". Int. J. Biol. Macromol. 34 (3): 203–11. doi:10.1016/j.ijbiomac.2004.04.003. PMID 15225993.
- ^ J. P. Gallivan; D. A. Dougherty (1999). "Cation–π interactions in structural biology". PNAS. 96 (17): 9459–9464. Bibcode:1999PNAS...96.9459G. doi:10.1073/pnas.96.17.9459. PMC 22230. PMID 10449714.
- ^ Burley, SK; Petsko, GA (1986). "Amino-aromatic interactions in proteins". FEBS Lett. 203 (2): 139–143. doi:10.1016/0014-5793(86)80730-X. PMID 3089835.
- ^ Brocchieri, L; Karlin, S (1994). "Geometry of interplanar residue contacts in protein structures". Proc. Natl. Acad. Sci. USA. 91 (20): 9297–9301. Bibcode:1994PNAS...91.9297B. doi:10.1073/pnas.91.20.9297. PMC 44799. PMID 7937759.
- ^ Karlin, S; Zuker, M; Brocchieri, L (1994). "Measuring residue associations in protein structures. Possible implications for protein folding". J. Mol. Biol. 239 (2): 227–248. doi:10.1006/jmbi.1994.1365. PMID 8196056.
- ^ Nandi, CL; Singh, J; Thornton, JM (1993). "Atomic environments of arginine side chains in proteins". Protein Eng. 6 (3): 247–259. doi:10.1093/protein/6.3.247. PMID 8506259.
- ^ Zhong, W; Gallivan, JP; Zhang, Y; Li, L; Lester, HA; Dougherty, DA (1998). "From ab initio quantum mechanics to molecular neurobiology: A cation–π binding site in the nicotinic receptor". Proc. Natl. Acad. Sci. USA. 95 (21): 12088–12093. Bibcode:1998PNAS...9512088Z. doi:10.1073/pnas.95.21.12088. PMC 22789. PMID 9770444.
- ^ D. L. Beene; G. S. Brandt; W. Zhong; N. M. Zacharias; H. A. Lester; D. A. Dougherty (2002). "Cation–π Interactions in Ligand Recognition by Serotonergic (5-HT3A) and Nicotinic Acetylcholine Receptors: The Anomalous Binding Properties of Nicotine". Biochemistry. 41 (32): 10262–9. doi:10.1021/bi020266d. PMID 12162741.
- ^ Xiu, Xinan; Puskar, Nyssa L.; Shanata, Jai A. P.; Lester, Henry A.; Dougherty, Dennis A. (2009). "Nicotine Binding to Brain Receptors Requires a Strong Cation–π Interaction". Nature. 458 (7237): 534–7. Bibcode:2009Natur.458..534X. doi:10.1038/nature07768. PMC 2755585. PMID 19252481.
- ^ Di Wu, W.; Hu, Q.; Yan, Z.; Chen, W.; Yan, C.; Huang, X.; Zhang, J.; Yang, P.; Deng, H.; Wang, J.; Deng, X.; Shi, Y. (2012). "Structural basis of ultraviolet-B perception by UVR8". Nature. 484 (7393): 214–219. Bibcode:2012Natur.484..214D. doi:10.1038/nature10931. PMID 22388820. S2CID 2971536.
- ^ Waksman, G; Kominos, D; Robertson, SC; Pant, N; Baltimore, D; Birge, RB; Cowburn, D; Hanafusa, H; et al. (1992). "Crystal structure of the phosphotyrosine recognition domain SH2 of v-src complexed with tyrosine-phosphorylated peptides". Nature. 358 (6388): 646–653. Bibcode:1992Natur.358..646W. doi:10.1038/358646a0. PMID 1379696. S2CID 4329216.
- ^ Wendt, K. U.; Poralla, K; Schulz, GE (1997). "Structure and Function of a Squalene Cyclase". Science. 277 (5333): 1811–5. doi:10.1126/science.277.5333.1811. PMID 9295270.
- ^ Aoki, K; K. Muyayama; H. Nishiyama (1995). "Cation–? interaction between the trimethylammonium moiety and the aromatic ring within lndole-3-acetic acid choline ester, a model compound for molecular recognition between acetylcholine and its esterase: an X-ray study". Journal of the Chemical Society, Chemical Communications (21): 2221–2222. doi:10.1039/c39950002221.
- ^ McCurdy, Alison; Jimenez, Leslie; Stauffer, David A.; Dougherty, Dennis A. (1992). "Biomimetic catalysis of SN2 reactions through cation–.pi. Interactions. The role of polarizability in catalysis". Journal of the American Chemical Society. 114 (26): 10314–10321. doi:10.1021/ja00052a031.
- ^ Fiedler (2005). "Selective Molecular Recognition, C-H Bond Activation, and Catalysis in Nanoscale Reaction Vessels". Accounts of Chemical Research. 38 (4): 351–360. CiteSeerX 10.1.1.455.402. doi:10.1021/ar040152p. PMID 15835881.
- ^ 애쉬튼, P. R., Goodnow, T. T., 카이퍼, A. E., M. V., 슬라윈, A. M. Z., 스펜서, N., 스토다트, J. F., 비센트, Ch. 그리고 윌리엄스, D. J. 안젤로. 화학. 인트. 1989년, 28년, 1396년–1399년 에드.
- ^ Knowles, Robert R.; Lin, Song; Jacobsen, Eric N. (2010). "Enantioselective Thiourea-Catalyzed Cationic Polycyclizations". Journal of the American Chemical Society. 132 (14): 5030–2. doi:10.1021/Ja101256v. PMC 2989498. PMID 20369901.
- ^ D. Quiñonero; C. Garau; C. Rotger; A. Frontera; P. Ballester; A. Costa; P. M. Deyà (2002). "Anion-π Interactions: Do They Exist?". Angew. Chem. Int. Ed. 41 (18): 3389–3392. doi:10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S.
- ^ P. de Hoog; P. Gamez; I. Mutikainen; U. Turpeinen; J. Reedijk (2004). "An Aromatic Anion Receptor: Anion-π Interactions do Exist". Angew. Chem. 116 (43): 5939–5941. doi:10.1002/ange.200460486.
원천
- J. C. Ma; D. A. Dougherty (1997). "The Cation–π Interaction". Chem. Rev. 97 (5): 1303–1324. doi:10.1021/cr9603744. PMID 11851453..
- Dougherty, D. A.; Stauffer, D. A. (Dec 1990). "Acetylcholine binding by a synthetic receptor: implications for biological recognition". Science. 250 (4987): 1558–1560. Bibcode:1990Sci...250.1558D. doi:10.1126/science.2274786. ISSN 0036-8075. PMID 2274786. S2CID 20210121.






{kind=link}