래디컬 중합
Radical polymerization프리라디칼 중합(FRP)은 프리라디칼 빌딩 블록을 연속적으로 추가하여 폴리머를 형성하는 폴리머 방식이다. 활성산소는 보통 별도의 이니시에이터 분자를 포함하는 여러 다른 메커니즘에 의해 형성될 수 있다. 그것의 세대에 따라, 개시 자유 급진주의자들은 (비방사성) 단량체를 더하고, 따라서 폴리머 체인을 성장시킨다.
프리라디칼 중합은 다양한 종류의 중합체와 재료 복합체를 얻기 위한 핵심 합성 경로다. 자유방사성 화학적 상호작용의 비교적 특수하지 않은 성질은 이것을 가장 다재다능한 형태의 중합체 중 하나로 만들고 고분자 자유방사성 사슬 끝단 및 다른 화학 물질이나 기판의 유연한 반응을 가능하게 한다. 2001년 미국에서 생산된 1,100억 파운드의 폴리머 중 400억 파운드는 프리라디칼 중합에 의해 생산되었다.[1]
프리라디칼 중합은 음이온, 양이온, 조정 중합과 함께 연쇄 성장 중합체의 일종이다.
입문
시작은 중합 과정의 첫 번째 단계다. 시작 중에 폴리머 체인이 생성되는 활성 중심이 생성된다. 모든 모노머가 모든 유형의 이니시에이터에 취약한 것은 아니다. 급진적 시작은 비닐 모노머의 탄소-탄소 이중 결합과 알데히드 및 케톤에서의 탄소-산소 이중 결합에 가장 효과적이다.[1] 입문에는 두 가지 단계가 있다. 첫 번째 단계에서, 하나 또는 두 개의 활성산소가 시작 분자에서 생성된다. 두 번째 단계에서는 활성산소가 이니시에이터 분자에서 존재하는 단량체로 전달된다. 이러한 개시자에 대해 몇 가지 선택사항을 이용할 수 있다.
시작 유형 및 이니시에이터
- 열분해
- 개시자는 결합이 균질하게 갈라져 두 개의 활성산소를 생성할 때까지 가열된다(그림 1). 이 방법은 유기 과산화물이나 아조 화합물에 가장 많이 사용된다.[3]
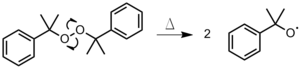 그림 1: 과산화디쿠밀의 열분해
그림 1: 과산화디쿠밀의 열분해 - 광분해
- 방사선은 균질적으로 결합을 분할하여 두 개의 활성산소를 생성한다(그림 2). 이 방법은 금속 요오드화물과 금속 알킬, 아조 화합물에 가장 많이 사용된다.[3] 광초기는 또한 과격파가 가장 낮은 3중 흥분 상태에 있을 때 2분자 H 추상화에 의해 발생할 수 있다.[4] 허용되는 광 이니시에이터 시스템은 다음 요구 사항을 충족해야 한다.[4]

- 리독스 반응
- 철에 의한 과산화수소 또는 과산화알킬 수소 감소([3]그림 3) Cr2+, V2+, Ti3+, Co2+, Cu와+ 같은 다른 환원제는 많은 경우에 철 이온 대신 사용될 수 있다.[1]
그림 3: 과산화수소와 철의 리독스 반응 |

- 페르설파테스
- 수성 단계에서 과황산염의 분해(그림 4). 이 방법은 에멀전 중합체에 유용하며, 에멀전 중합체에서는 급진성이 소수성 단량체 함유 방울로 확산된다.[3]
- 전리방사선
- α-, β-, γ- 또는 x-선은 시작 종에서 전자가 방출되는 원인이 되며, 그 다음에 분열과 전자 포획이 일어나 급진성을 생성한다(그림 5).[3]
- 전기화학
- 모노머와 전해질을 모두 함유한 용액의 전기분해. 모노머 분자는 음극에서 전자를 받아 급진 음이온이 되고, 모노머 분자는 양극에서 전자를 포기해 급진적인 양이온이 된다(그림 6). 그러면 급진 이온은 자유 급진 중합(및/또는 이온)을 시작한다. 이러한 유형의 시작은 특히 폴리머 필름으로 금속 표면을 코팅하는 데 유용하다.[5]
- 플라즈마
- 기체 모노머는 플라즈마(이온화된 기체 분자)가 생성되는 조건에서 낮은 압력에서 전기 방전에 배치된다. 어떤 경우에는 시스템을 가열하거나 무선 주파수 장에 배치하여 플라즈마 생성을 돕는다.[1]
- 소닉
- 인체 청력 범위(16kHz) 이상의 주파수에서 고강도 초음파를 모노머에 적용할 수 있다. 시작은 공동화 효과(액체 내 공동의 형성 및 붕괴)에서 비롯된다. 충치의 붕괴는 매우 높은 국소 온도와 압력을 발생시킨다. 이로 인해 흥분한 전자 상태가 형성되고, 이는 결국 채권 파괴와 급진적인 형성으로 이어진다.[1]
- 3차 이니시에이터
- 3차 이니시에이터는 여러 유형의 이니시에이터를 하나의 개시 시스템에 결합하는 것이다. 개시자의 유형은 자신이 생산하는 중합체에서 유도하는 것으로 알려진 성질에 기초하여 선택된다. 예를 들어 폴리(메틸메타크릴레이트)는 과산화지질 벤조일-3,6-bis(o-카르복시벤조일)-N-이소프로필카르바졸-di---indenylzicronium5 디클로로이드(그림 7)[6][7]에 의해 합성되었다. 이러한 유형의 개시 시스템에는 메탈로세, 이니시에이터, 그리고 이단자체디케토카르복실산이 포함되어 있다. 메탈로케인은 개시자와 결합하여 폴리(메틸메타크릴레이트)의 중합성을 가속화하고 분자량 분포가 좁은 폴리머를 생성한다. 여기에 제시된 예는 인데닐지르코늄(Metalocene)과 과산화벤조일(Benzoyl)로 구성되어 있다. 또한 이 예에서 3,6-bis(o-carboxybenzoyl)-N-isopropylcarbazole과 같이 이단성 디케토 카르복실산을 포함하는 개시 시스템은 과산화 벤조일 분해에 촉매 작용을 하는 것으로 알려져 있다. 이 특별한 이단성 디케트 카르복실산을 사용한 시스템을 시작하는 것은 또한 폴리머의 미세 구조에 영향을 미치는 것으로 알려져 있다. 이 모든 구성 요소들 - 금속성, 이니시에이터, 그리고 이단성 디케토 카르복실산 -의 조합은 중합 속도를 가속화하고 강화된 내열성과 규칙적인 미세 구조를 가진 중합체를 생성하는 것으로 보여지는 3차 개시 시스템을 제공한다.[6][7]




이니시에이터 효율
과격한 종에 대한 부작용과 비효율적인 합성 때문에 체인 개시는 100%가 [clarify]아니다. 효율 계수 f는 효과적인 급진적 집중을 설명하기 위해 사용된다. f의 최대값은 1이지만, 대표적인 값은 0.3에서 0.8까지이다. 다음은 이니시에이터의 효율을 떨어뜨리는 반응 목록이다.
- 일차 재조합
- 두 개의 활성산소가 체인을 시작하기 전에 재결합한다(그림 8). 이것은 용제 케이지 안에서 발생하는데, 이는 새로운 활성산소 사이에 용제가 아직 오지 않았다는 것을 의미한다.[3]

- 사이드 리액션
- 생산할 수 있는 세 가지 급진성 대신에 하나의 급진성이 생성된다(그림 10).[3]
그림 10: 폴리머 체인 R과 다른 종의 반응 |

전파
중합하는 동안, 폴리머는 그것의 대부분의 시간을 그것의 체인 길이를 늘리거나, 또는 전파하는 데 보낸다. 급진적 이니시에이터가 형성되면 모노머를 공격한다(그림 11).[8] 에테네 단량체에서는 시그마 결합으로 두 탄소 사이에 하나의 전자쌍이 단단히 고정되어 있다. 다른 하나는 파이 본드에 더 느슨하게 고정되어 있다. 프리 래디컬은 파이 결합에서 나온 하나의 전자를 사용하여 탄소 원자와의 보다 안정적인 결합을 형성한다. 다른 전자는 두 번째 탄소 원자로 돌아와 분자 전체를 또 다른 급진적인 것으로 바꾼다. 이것은 폴리머 체인을 시작한다. 그림 12는 에틸렌 단량체의 궤도가 급진적 이니시에이터와 어떻게 상호작용하는지를 보여준다.[9]


체인이 일단 시작되면 체인은 더 이상 모노머(살아있는 중합)가 없거나 종료될 때까지 전파된다(그림 13). 급진적 및 연쇄 반응성, 용매 및 온도 등의 여러 요인에 따라 몇 개에서 수천 개까지의 전파 단계가 있을 수 있다.[10][11] 체인 전파의 메커니즘은 다음과 같다.

종료
급진적인 중합에서는 활성산소의 높은 반응성으로 인해 연쇄 해지가 불가피하다. 종단은 몇 가지 다른 메커니즘에 의해 발생할 수 있다. 더 긴 체인을 원하는 경우 이니시에이터 농도를 낮게 유지해야 하며 그렇지 않으면 많은 짧은 체인이 발생할 수 있다.[3]
- 두 개의 활성 체인 끝의 조합: 다음 프로세스 중 하나 또는 둘 다 발생할 수 있다.
- 활성 체인 끝과 이니시에이터 래디컬의 결합(그림 16).[3]
- 불순물 또는 억제제와의 상호작용. 산소는 일반적인 억제제다. 성장 사슬은 분자 산소와 반응하여 훨씬 덜 반응하는 산소 레디컬을 생성한다(그림 17). 이것은 전파 속도를 현저하게 늦춘다. 니트로벤젠, 부틸화 하이드록실 톨루엔, 디페닐 피크릴 하이드레이질(DPPH, 그림 18)은 몇 가지 다른 억제제들이다. 후자는 과격파의 공명이 안정되기 때문에 특히 효과적인 억제제다.[3]





체인 전이
다른 종단 방식과 달리 체인 전환은 단 하나의 급진주의자를 파괴하는 결과를 낳지만 또 다른 급진주의자를 창조하는 결과를 낳는다. 그러나 종종 새롭게 만들어진 이 급진주의자는 더 이상 전파할 수 없다. 불균형과 유사하게, 모든 체인 전달 메커니즘은 수소나 다른 원자의 추상화를 포함한다. 체인 전송 메커니즘에는 몇 가지 유형이 있다.[3]
- 용매: 수소 원자는 용매 분자에서 추출되어 용매 분자에 급진적인 형성을 초래하며, 이는 더 이상 전파되지 않는다(그림 19). 용제 분자를 포함하는 체인 전달의 효과는 존재하는 용매의 양(용매 리드가 많을수록 전달 확률이 높아짐), 추상화 단계에 관여하는 결합의 강도(용제 결합은 전달 확률이 커짐), 형성되는 용매 래디컬의 안정성(더 큰 안정성으로 연결됨)에 따라 달라진다.y는 더 큰 전이 확률을 유도한다). 불소를 제외한 할로겐은 쉽게 옮겨진다.[3]
- 단량체에게: 수소 원자는 단량체에서 추출된다. 이것이 영향을 받은 모노머에 대해 급진적인 것을 만들어내지만, 이 급진적인 모노머의 공명 안정화는 더 이상의 전파를 방해한다(그림 20).[3]
- 이니시에이터에게: 폴리머 체인은 이니시에이터와 반응하며, 이는 그 폴리머 체인을 종료하지만 새로운 래디컬 이니시에이터를 생성한다(그림 21). 이 이니시에이터는 새로운 폴리머 체인을 시작할 수 있다. 그러므로 다른 형태의 체인 전송과는 달리, 이니시에이터로의 체인 전송은 추가 전파를 허용한다. 과산화수소 개시자는 체인 전송에 특히 민감하다.[3]
- 고분자: 고분자 사슬의 래디컬은 다른 고분자 사슬의 어딘가에서 수소 원자를 추상화한다(그림 22). 이것은 하나의 폴리머 체인의 성장을 종식시키지만, 다른 하나는 가지를 치고 성장을 재개할 수 있게 한다. 이 반응 단계는 중합체 체인의 수나 중합된 모노머의 수를 변경하지 않으므로, 중합 평균의 수치에 영향을 미치지 않는다.[12]
체인 전송의 영향: 체인 전달의 가장 분명한 효과는 폴리머 체인 길이의 감소다. 전달 속도가 전파 속도보다 훨씬 큰 경우, 2-5개의 반복 단위(단층화)의 체인 길이를 가진 초소형 폴리머가 형성된다.[13] The Mayo equation estimates the influence of chain transfer on chain length (xn): 여기서 k는tr 체인 전송에 대한 속도 상수이고 k는p 전파에 대한 속도 상수다. Mayo 방정식은 용매로의 이전이 주요한 종단 경로라고 가정한다.[3][14]
![{\frac {1}{x_{n}}}=\left({\frac {1}{x_{n}}}\right)_{o}+{\frac {k_{{tr}}[solvent]}{k_{p}[monomer]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3af10ecbdc45b856a8cd76ecc9502f63fc6bdc03)
방법들
급진적 중합에는 네 가지 산업적 방법이 있다.[3]
- 벌크 중합: 반응 혼합물은 이니시에이터와 단량체만 포함하고 용제는 없다.
- 용액 중합: 반응 혼합물은 용매, 이니시에이터, 단량체를 포함한다.
- 서스펜션 중합: 반응 혼합물은 수용성 단층체, 수용성 단층체 및 단층체 방울에 용해되는 이니시에이터를 포함한다(단층체와 이니시에이터 모두 소수성).
- 에멀전 중합화: 단량체 방울이 아닌 수용성 단계에 이니시에이터가 용해된다는 점을 제외하면 서스펜션 중합화와 유사하다(단수체는 소수성, 이니시에이터는 소수성). 유화제 또한 필요하다.
그 밖의 급진적 중합 방법으로는 다음과 같은 것이 있다.
- 템플릿 중합: 이 과정에서, 폴리머 체인은 일생 중 많은 기간 동안 고분자를 따라 자랄 수 있다. 잘 조화된 템플릿은 중합률뿐만 아니라 딸 폴리머의 어금니 질량과 미세 구조에 영향을 미칠 수 있다. 딸 폴리머의 어금니 질량은 템플릿이 없을 때 생성된 폴리머의 질량보다 최대 70배까지 클 수 있으며, 어금니 질량이 템플릿 자체보다 높을 수 있다. 이는 템플릿 관련 활성산소에 대한 종단 지연과 템플릿 폴리머의 끝에 도달한 후 인접한 템플릿에 래디컬을 홉으로써 발생하기 때문이다.[15]
- 혈장 중합: 중합은 플라즈마로 시작한다. 알케인, 알케인, 알카인을 포함한 다양한 유기 분자는 이러한 조건에서 고분자량 생산물에 중합된다. 번식 메커니즘은 이온종과 급진종을 모두 포함하는 것으로 보인다. 플라즈마 중합은 박막 콘덴서, 편향 방지 코팅 및 다양한 유형의 박막과 같은 용도에 사용할 수 있는 잠재적으로 독특한 방법을 제공한다.[1]
- 소닉: 중합은 고강도 초음파에 의해 시작된다. 고분자 중량 중합체에 대한 중합이 관찰되지만 변환은 낮다(<15%). 중합은 낮은 변환에서도 발생하는 높은 점도로 인해 자가 제한된다. 점성이 높으면 충동과 급진적인 생산을 방해한다.[1]
가역적 비활성화 급진적 중합
살아있는 급진적 중합, 제어된 급진적 중합, 가역적 비활성화 급진적 중합(RDRP)은 불순물에 의한 종료를 방지하는 완전한 순수 반응에 의존한다. 이러한 중합은 더 이상의 단량체가 없을 때만 멈추기 때문에, 더 많은 단량체가 추가될 때 중합은 계속될 수 있다. 블록 복합체는 이런 식으로 만들어질 수 있다. RDRP는 분자량 및 분산 제어를 허용한다. 그러나 이것은 달성하기가 매우 어렵고 대신 분자량 및 분산에 대한 부분적인 제어만으로 사이비 생물 중합이 일어난다.[15] ATRP와 RAFT는 완전한 급진적 중합화의 주요 유형이다.
- 원자 전달 급진 중합(ATRP): 원자 전달 급진적 첨가물에 의한 탄소-탄소 결합 형성에 기초한다. 1995년 사와모토[16] 미쓰오, 왕진산토프, 마티야스체프스키가 독자적으로 발견한 이 방법은 휴면종(알킬 할라이드 등)과 전이금속 할라이드 촉매(휴면종 활성화)의 가역 활성화가 필요하다.[17][18][3]
- 가역성 첨가-파편성 체인-전송 중합체(RAFT): 디티오 화합물과 같이 가역성 체인 전달제 역할을 할 수 있는 화합물이 필요하다.[3]
- 안정적 자유 래디컬 폴리머화(SFRP): 각 폴리머 체인에 분자량 분포가 좁은 선형 또는 분기형 폴리머와 반응형 엔드 그룹을 합성하는 데 사용된다. 이 과정은 또한 독특한 특성을 가진 블록 공동 폴리머를 만드는 데 사용되었다. 변환 속도는 이 공정을 사용하여 약 100%이지만 약 135 °C의 온도가 필요하다. 이 과정은 아크릴레이트, 스타일렌, 디엔과 함께 가장 일반적으로 사용된다. 그림 23의 반응 체계는 SFRP 과정을 보여준다.[19] 체인 엔드는 TEMO 분자(그림 24)로 기능하기 때문에 커플링에 의한 조기 종단이 감소한다. 살아있는 모든 중합체와 마찬가지로, 폴리머 체인은 모든 모노머가 소비될 때까지 성장한다.[19]


키네틱스
일반적인 연쇄 성장 중합에서 시작, 전파 및 종료에 대한 반응 속도는 다음과 같이 설명할 수 있다.
![v_{i}={\operatorname {d}[M\cdot ]/\operatorname {d}t}=2k_{d}f[I]](https://wikimedia.org/api/rest_v1/media/math/render/svg/f02573160d99ce28cd15dfe6c9f111e9c47c082d)
![v_{p}=k_{p}[M][M\cdot ]](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc7bb155b647c8e9a58d7d300e71a52898b8f221)
![v_{t}={-\operatorname {d}[M\cdot ]/\operatorname {d}t}=2k_{t}[M\cdot ]^{2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e451f440b60e6b1e9fbe627b99a0d6eb734aeea4)
여기서 f는 이니시에이터의 효율이며 kd, kp, k는t 각각 이니시에이터의 분리, 체인 전파, 종단 등에 대한 상수다. [I] [M]과 [M•]은 이니시에이터, 모노머 및 활성 성장 체인의 농도다.
정상 상태 근사치에서 활성 성장 사슬의 농도는 일정하게 유지된다. 즉, 시작 속도와 종료 속도는 동일하다. 활성 사슬의 농도는 시스템에서 다른 알려진 종에 의해 도출되고 표현될 수 있다.
![[M\cdot ]=\left({\frac {k_{d}[I]f}{k_{t}}}\right)^{{1/2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/92af6e128be7fa6a566a3590c6f267e601e5e3e2)
이 경우, 체인 전파 속도는 이니시에이터와 모노머 농도의[20][21] 함수를 사용하여 더 자세히 설명할 수 있다.
![{\displaystyle v_{p}={k_{p}}\left({\frac {fk_{d}}{k_{t}}}\right)^{1/2}[I]^{1/2}[M]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8fab2227059300c21a6173d5108ca221645da4cf)
운동 체인 길이 v는 수명 동안 활성 중심과 반응하는 모노머 단위의 평균 수를 측정한 것으로 종단 메커니즘을 통해 분자량과 관련이 있다. 체인 전송이 없다면, 운동 체인 길이는 전파율과 개시율의 함수일 뿐이다.[22]
![{\displaystyle \nu ={\frac {v_{p}}{v_{i}}}={\frac {k_{p}[M][M\cdot ]}{2fk_{d}[I]}}={\frac {k_{p}[M]}{2(fk_{d}k_{t}[I])^{1/2}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/60318ebbc0960f885949ea451e3692f7cebae026)
반응에서 체인 전달 효과가 발생하지 않는다고 가정할 때, 중합 P의n 수 평균 정도는 운동 체인 길이와 상관될 수 있다. 불균형에 의한 종단의 경우, 모든 운동 체인당 하나의 폴리머 분자가 생성된다.

조합에 의한 종단은 두 개의 운동 체인당 하나의 폴리머 분자로 이어진다.[20]

이 두 메커니즘의 혼합은 전체 종료 프로세스에 대한 불균형의 기여인 값을 사용하여 설명할 수 있다.

체인 이전을 고려한다면, 비록 다중 폴리머 체인이 생성되지만, 개시 단계에 의해 생성되는 자유방사선 중심은 어떤 체인 이체 사건 이후에도 존속하기 때문에 운동 체인 길이는 전달 과정의 영향을 받지 않는다. 그러나 체인이 이동함에 따라 평균 중합 정도가 감소하는데, 이는 체인이 이동함에 따라 성장하는 체인이 체인 전송 이벤트에 의해 종료되기 때문이다. 용제 S, 이니시에이터 I, 폴리머 P 및 추가된 체인 전달 물질 T에 대한 체인 전달 반응을 고려한다. P의n 방정식은 다음과 같이 수정된다.[23]
![{\displaystyle {\frac {1}{x_{n}}}={\frac {2k_{t,d}+k_{t,c}}{{k_{p}}^{2}[M]^{2}}}v_{p}+C_{M}+C_{S}{\frac {[S]}{[M]}}+C_{I}{\frac {[I]}{[M]}}+C_{P}{\frac {[P]}{[M]}}+C_{T}{\frac {[T]}{[M]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f87872861546ef6fc87a203d04693d6898ca25d2)
다른 분자에 대해 체인 전달 상수 C를 정의하는 것은 보통이다.
- , , C = k { = t t t }}}, C = k t = t t t p{\p}{p}}




열역학
연쇄 성장 중합에서 중합체와 단량체 사이의 평형 위치는 중합체의 열역학으로 결정할 수 있다. 중합체의 Gibbs 자유 에너지(ΔGp)는 일반적으로 중합체 반응의 경향을 정량화하는 데 사용된다. ΔGp < 0이면 중합이 선호되고, ΔGp > 0이면 중합체가 탈고된다. 열역학 방정식 ΔG = ΔH – ΔS에 따르면 음의 엔탈피와 증가하는 엔트로피가 중합으로 평형을 이동시킨다.
일반적으로 중합체는 발열 과정, 즉 음의 엔탈피 변화로, 성장하는 중합체 체인에 모노머를 추가하는 것은 π 본드를 bonds 본드로 변환하거나, 고리 장력을 순환 단량체로 방출하는 고리 개방 반응을 수반하기 때문이다. 한편 중합하는 동안 다량의 작은 분자가 연관되어 회전과 변환의 자유도가 상실된다. 그 결과 거의 모든 중합 공정에서 엔트로피는 시스템 ΔSp < 0이 감소한다. ΔH는p 거의 항상 엔트로피적으로 선호되기 때문에 ΔH는 충분히 음수가 되어야 비우호적 항을 보상할 수 있다. 그래야만 중합이 음의 ΔG에p 의해 열역학적으로 선호될 것이다.
실제로 중합은 저온에서 선호된다. ΔS는p 작다. 고온에서는 고열화를 선호한다. ΔS는p 크다. 온도가 상승할수록 ΔG는p 음수가 감소한다. 일정 온도에서 중합은 평형(중합률 = 중합률)에 도달한다. 이 온도를 천장 온도(Tc)라고 한다. ΔGp = 0.[24]
입체화학
중합체의 입체화학은 화학적 구성이 동일한 중합체의 원자연결성과 공간지향성의 차이와 관련이 있다.
헤르만 스토딩거는 1920년대 후반에 비닐 단층체의 연쇄 중합에 있어서 입체파를 연구했고, 폴리머 성장의 각각의 전파 단계가 입체파를 일으킬 수 있다는 생각을 사람들이 충분히 이해하는데 20년이 더 걸렸다. 입체화학에서 중요한 이정표는 1950년대 지글러와 나타, 그리고 그들의 동료들이 입체 중합체를 합성하기 위한 금속 기반 촉매를 개발하면서 확립되었다. 폴리머의 입체화학이 특히 관심을 갖는 이유는 폴리머의 물리적 거동은 일반적인 화학적 구성뿐만 아니라 미세 구조의 보다 미묘한 차이에도 좌우되기 때문이다.[25] 아틀라스틱 폴리머는 스테레오 화학의 무작위 배열로 구성되며, 아모르퍼스(비결정질)로, 체력이 낮은 부드러운 물질이다. 해당 동위원소(동일한 측면의 모든 대체물과 같은)와 신디오토틱(동일한 측면의 대체 반복 장치의 대체물과 같은) 중합체는 대개 결정성이 높은 물질로서 얻는다. 입체 중합체는 순서가 더 높고 그 결과 결정성이 체력을 높이고 용매와 화학 저항성뿐만 아니라 결정성에 의존하는 다른 성질의 차이로 이어지기 때문에 수정 격자로 포장하는 것이 더 쉽다. 입체 폴리머의 산업 효용성의 대표적인 예가 폴리프로펜이다. 동위원소 폴리프로펜은 고융화(165 °C), 강력하고 결정성이 강한 폴리머로 플라스틱과 섬유로 모두 사용된다. 아틀락틱 폴리프로펜은 윤활제, 실란트, 접착제 등에 사용되는 아스팔트 혼합과 제형에 사용되는 기름기가 많고 왁스 같은 부드러운 외관을 가진 비정형 물질이지만, 동위원소 폴리프로펜에 비해 부피는 미미하다.
모노머가 급진적인 체인 끝에 추가될 때, 그것의 입체화학성과 관련하여 고려해야 할 두 가지 요소가 있다: 1) 단자 체인 탄소와 접근하는 모노머 분자 사이의 상호작용, 2) 폴리머 체인에서 음각 반복 유닛의 구성.[5] 단자 탄소 원자는 sp2 하이브리드를 가지고 있으며 평면이다. 모노머 CH2=CXY의 중합성을 고려한다. 모노머 분자가 단자 탄소에 접근할 수 있는 두 가지 방법이 있다: 거울 접근법(동일한 측면에 유사한 대체물이 있음)과 비거울 접근법(동일한 측면에 대체물이 있음)이다. 다음 모노머가 추가되기 전에 자유 회전이 발생하지 않는 경우, 미러 접근방식은 항상 동위원소 중합체로 이어지고 비거울 접근방식은 항상 신디오토틱 중합체로 이어진다(그림 25).[5]

단, Penultimate 반복 유닛과 단자 탄소 원자의 대체물 사이의 교호작용이 유의한 경우, 정전기적 교호작용을 최소화하는 방식으로 모노머를 폴리머에 추가할 수 있다(그림 26).[5]

반응도
전통적으로, 단량체와 활성산소의 반응성은 복합화 데이터를 통해 평가된다. 단량체 반응률 비율의 반정량적 예측을 위해 가장 널리 사용되는 도구인 Q-e 체계는 1947년 Alfrey와 Price에 의해 처음 제안되었다.[26] 이 계획은 전환 상태에서 본질적인 열역학적 안정성과 극지 효과를 고려한다. 주어진 래디컬 과(와) M j {\{j은 각각 내인성 P와i Q를j 갖는 것으로 간주된다.[27] 전환 상태의 극지방 효과, 즉 그 실체가 운반하는 것으로 추정되는 영구 전기 전하(라디칼 또는 분자)는 주어진 모노머에 대한 상수인 e 인자에 의해 정량화되며, 그 특정 모노머에서 파생된 급진자에 대해서도 동일한 값을 갖는다. 활성 엔드가 모노머 1의 래디컬인 성장하는 폴리머 체인에 모노머 2를 추가하기 위해, 속도 상수 k는 다음과12 같은 4개의 관련 반응도 파라미터와 관련이 있다고 가정한다.
 (와)
(와) 

이 체인에 모노머 1과 2를 추가하기 위한 모노머 반응도 비율은 다음과[27][28] 같다.

주어진 쌍의 모노머의 복합화의 경우, 두 실험 반응도 비율 r과1 r은2 (Q1/Q2)와 (e1 – e2)의 평가를 허용한다. 그런 다음 각 모노머에 대한 값을 참조 모노머에 상대적으로 할당할 수 있으며, 일반적으로 임의의 값 Q = 1.0 및 e = –0.8과 함께 스티렌으로 선택된다.[28]
적용들
프리 래디컬 중합은 폴리스티렌, 열가소성 플라스틱 블록 복합체 엘라스토머,[29] 심혈관계 스텐트,[30] 화학 계면활성제[31] 및 윤활제의 제조를 포함한 용도를 발견했다. 블록 겸용기는 접착제, 신발, 장난감 등 다양한 용도로 사용된다.
자유 급진적 중합은 탄소 나노튜브의 기능화와 같은 연구에서도 사용된다.[32] CNT의 내재적 전자 속성은 그들이 솔루션에서 대형 집계를 형성하도록 유도하여 유용한 응용 프로그램을 배제한다. CNT의 벽에 작은 화학 그룹을 추가하면 이러한 성향을 없애고 주변 환경에 대한 반응을 조절할 수 있다. 작은 분자 대신 중합체를 사용하면 CNT 특성을 수정할 수 있다(반대로 나노튜브는 중합체의 기계적, 전자적 특성을 수정할 수 있다).[29] 예를 들어 연구진은 먼저 연쇄 래디컬 중합으로 폴리스티렌을 중합한 뒤 130℃에서 탄소 나노튜브와 혼합해 활성산소를 만들어 탄소 나노튜브 벽에 접목시켜 탄소 나노튜브를 코팅했다(그림 27).[33] 연쇄 성장 중합("접종 대상")은 미리 결정된 성질을 지닌 중합체를 합성한다. 폴리머의 정화는 접붙이기 전에 보다 균일한 길이 분포를 얻기 위해 사용될 수 있다. 반대로 원자전달중합화(ATRP)나 니트로크사이드 매개중합화(NMP)와 같은 급진적 중합화 기법으로 "접종"하면 고분자중량 중합체의 급속한 성장이 가능하다.

또한 급진적인 중합은 나노콤포사이트 하이드로겔의 합성을 돕는다.[34] 이 겔들은 네트워크 폴리머에 싸인 물에 잘 녹는 나노 스케일의 점토(특히 스몰테이트로 분류된 점토)로 만들어진다. 그것들은 종종 생체 적합성이 있고 합성 조직과 같은 응용을 약속하는 기계적 특성(유연성과 강도 등)을 가지고 있다. 합성은 자유로운 급진적 중합성을 포함한다. 일반적인 합성 절차는 그림 28에 설명되어 있다. 점토는 물에 분산되어 매우 작고 다공성 판을 형성한다. 다음으로 이니시에이터와 촉매가 추가되고 그 다음에는 유기 모노머, 즉 일반적으로 아크릴아미드 또는 아크릴아미드 파생물이 추가된다. 개시자는 유기 모노머보다 점토와의 상호작용이 강하도록 선택되어 있어 점토 표면에 우선적으로 흡착한다. 혼합물과 물 용제를 가열하여 중합작용을 시작한다. 중합체는 점토에 차례로 묶여 있는 개시자를 떼어낸다. 재조합과 불균형 반응으로 인해 성장하는 폴리머 체인은 서로 결합하여 강력한 교차 연계 네트워크 폴리머를 형성하며, 점토 입자가 다중 폴리머 체인 세그먼트의 분기점 역할을 한다.[35] 이러한 맥락에서 사용되는 자유 급진적 중합은 다양한 기질(적합한 클라이의 화학자는 다양하다)에서 중합체의 합성을 가능하게 한다. 연쇄 성장 중합에 고유한 종단 반응은 유연성, 기계적 강도 및 생체적합성을 가진 물질을 생성한다.

전자제품
래디컬 폴리머 글라스 PTMA는 일반적인 반도체용 폴리머에 비해 전기 전도성이 약 10배 높다. PTMA는 투명한 태양전지, 이동전화 디스플레이를 위한 항정전기 및 항원기 코팅, 낙뢰로부터 보호하기 위한 항공기용 항정전기 커버, 유연한 플래시 드라이브, 전기를 열과 반대로 변환하는 열전 소자에 사용될 수 있는 전기 활성 고분자 종류에 속한다. 광범위한 실용적 응용은 전도성을 100배에서 1,000배 더 증가시켜야 한다.
이 중합체는 펜던트 그룹의 특정 수소 원자를 산소 원자로 교체하는 것을 포함하는 제독법을 사용하여 만들어졌다. PTMA에서 발생하는 산소 원자는 외부 껍질 안에 한 개의 전자가 손상되지 않아 전하 운반이 용이하다. 방제 단계는 네 가지 뚜렷한 화학적 기능성으로 이어질 수 있으며, 그 중 두 가지는 전도성을 증가시킬 수 있다.[36]
참고 항목
참조
- ^ a b c d e f g Odian, George (2004). Principles of Polymerization (4th ed.). New York: Wiley-Interscience. ISBN 978-0-471-27400-1.
- ^ Jenkins, A. D.; Kratochvíl, P.; Stepto, R. F. T.; Suter, U. W. (1996). "Glossary of basic terms in polymer science (IUPAC Recommendations 1996)" (PDF). Pure and Applied Chemistry. 68 (12): 2287–2311. doi:10.1351/pac199668122287.
- ^ a b c d e f g h i j k l m n o p q r s Cowie, J. M. G.; Arrighi, Valeria (2008). Polymers: Chemistry and Physics of Modern Materials (3rd ed.). Scotland: CRC Press. ISBN 978-0-8493-9813-1.
- ^ a b Hageman, H. J. (1985). "Photoinitiators for Free Radical Polymerization". Progress in Organic Coatings. 13 (2): 123–150. doi:10.1016/0033-0655(85)80021-2.
- ^ a b c d e Stevens, Malcolm P. (1999). Polymer Chemistry: An Introduction. New York: Oxford University Press. ISBN 978-0-19-512444-6.
- ^ a b Islamova, R. M.; Puzin, Y. I.; Kraikin, V. A.; Fatykhov, A. A.; Dzhemilev, U. M. (2006). "Controlling the Polymerization of Methyl Methacrylate with Ternary Initiating Systems". Russian Journal of Applied Chemistry. 79 (9): 1509–1513. doi:10.1134/S1070427206090229.
- ^ a b Islamova, R. M.; Puzin, Y. I.; Fatykhov, A. A.; Monakov, Y. B. (2006). "A Ternary Initiating System for Free Radical Polymerization of Methyl Methacrylate". Polymer Science, Series B. 48 (3): 130–133. doi:10.1134/S156009040605006X.
- ^ "Addition Polymerization". Materials World Modules. June 2009. Retrieved 1 April 2010.
- ^ a b "Polymer Synthesis". Case Western Reserve University. 2009. Archived from the original on 7 February 2010. Retrieved 10 March 2010.
- ^ Leach, Mark R. "Radical Chemistry". Chemogenesis. Retrieved 2 April 2010.
- ^ Pojman, John A.; Jason Willis; Dionne Fortenberry; Victor Ilyashenko; Akhtar M. Khan (1995). "Factors affecting propagating fronts of addition polymerization: Velocity, front curvature, temperature profile, conversion, and molecular weight distribution". Journal of Polymer Science Part A: Polymer Chemistry. 33 (4): 643–652. Bibcode:1995JPoSA..33..643P. doi:10.1002/pola.1995.080330406.
- ^ 루딘, 알프레드 폴리머 이공학의 요소 (Acadical Press 1982년) 페이지 220 ISBN 0-12-601680-1
- ^ 루딘, 알프레드 폴리머 이공학의 요소 (Academic Press 1982년) p.212 ISBN 0-12-601680-1
- ^ 체인 이전을 위한 메이요 방정식은 복합체를 위한 메이요-르위스 방정식과 혼동해서는 안 된다.
- ^ a b Colombani, Daniel (1997). "Chain-Growth Control in Free Radical Polymerization". Progress in Polymer Science. 22 (8): 1649–1720. doi:10.1016/S0079-6700(97)00022-1.
- ^ Kato, M; Kamigaito, M; Sawamoto, M; Higashimura, T (1995). "Polymerization of Methyl Methacrylate with the Carbon Tetrachloride / Dichlorotris-(triphenylphosphine)ruthenium(II) / Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization". Macromolecules. 28 (5): 1721–1723. Bibcode:1995MaMol..28.1721K. doi:10.1021/ma00109a056.
- ^ Wang, J-S; Matyjaszewski, K (1995). "Controlled/"living" radical polymerization. Atom transfer radical polymerization in the presence of transition-metal complexes". J. Am. Chem. Soc. 117 (20): 5614–5615. doi:10.1021/ja00125a035.
- ^ "The 2011 Wolf Prize in Chemistry". Wolf Fund. Retrieved 21 February 2011.
- ^ a b "Stable Free Radical Polymerization". Xerox Corp. 2010. Archived from the original on 28 November 2003. Retrieved 10 March 2010.
- ^ a b Cowie, J. M. G. (1991). Polymers: Chemistry and Physics of Modern Materials (2nd ed.). Blackie (USA: Chapman & Hall). pp. 58–60. ISBN 978-0-216-92980-7.
- ^ 루딘, 알프레드 폴리머 이공학의 요소 (Academic Press 1982년) pp.195-9 ISBN 0-12-601680-1
- ^ 루딘, 알프레드 폴리머 이공학의 요소 (Academic Press 1982년) p.209 ISBN 0-12-601680-1
- ^ 루딘, 알프레드 폴리머 이공학의 요소 (Academic Press 1982년) p.214 ISBN 0-12-601680-1
- ^ 튀긴 조엘 R. 폴리머 과학기술 (제2편, 프렌티스홀 2003) 페이지 39 ISBN 0-13-018168-4
- ^ Clark, Jim (2003). "The Polymerization of Alkenes". ChemGuide. Retrieved 1 April 2010.
- ^ Alfrey, Turner; Price, Charles C. (1947). "Relative reactivities in vinyl copolymerization". Journal of Polymer Science. 2 (1): 101–106. Bibcode:1947JPoSc...2..101A. doi:10.1002/pol.1947.120020112.
- ^ a b Allcock H.R, Lampe F.W., Mark J.E. 컨템포러리 폴리머 화학 (3차 에드워드, Pearson Fatherice-Hall 2003) p.364 ISBN 0-13-065056-0
- ^ a b 루딘, 알프레드 폴리머 이공학의 요소 (Academic Press 1982년) 페이지 289 ISBN 0-12-601680-1
- ^ a b Braunecker, W. A.; K. Matyjaszewski (2007). "Controlled/living radical polymerization: Features, developments, and perspectives". Progress in Polymer Science. 32 (1): 93–146. doi:10.1016/j.progpolymsci.2006.11.002.
- ^ Richard, R. E.; M. Schwarz; S. Ranade; A. K. Chan; K. Matyjaszewski; B. Sumerlin (2005). "Evaluation of acrylate-based block copolymers prepared by atom transfer radical polymerization as matrices for paclitaxel delivery from coronary stents". Biomacromolecules. 6 (6): 3410–3418. doi:10.1021/bm050464v. PMID 16283773.
- ^ Burguiere, C.; S. Pascual; B. Coutin; A. Polton; M. Tardi; B. Charleux; K. Matyjaszewski; J. P. Vairon (2000). "Amphiphilic block copolymers prepared via controlled radical polymerization as surfactants for emulsion polymerization". Macromolecular Symposia. 150: 39–44. doi:10.1002/1521-3900(200002)150:1<39::AID-MASY39>3.0.CO;2-D.
- ^ Homenick, C. M.; G. Lawson; A. Adronov (2007). "Polymer grafting of carbon nanotubes using living free-radical polymerization". Polymer Reviews. 47 (2): 265–270. doi:10.1080/15583720701271237.
- ^ Lou, X. D.; C. Detrembleur; V. Sciannamea; C. Pagnoulle; R. Jerome (2004). "Grafting of alkoxyamine end-capped (co)polymers onto multi-walled carbon nanotubes". Polymer. 45 (18): 6097–6102. doi:10.1016/j.polymer.2004.06.050.
- ^ Haraguchi, K. (2008). "Nanocomposite hydrogels". Current Opinion in Solid State and Materials Science. 11 (3–4): 47–54. Bibcode:2007COSSM..11...47H. doi:10.1016/j.cossms.2008.05.001.
- ^ Haraguchi, K.; Takehisa T. (2002). "Nanocomposite hydrogels: a unique organic-inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties". Advanced Materials. 14 (16): 1120–1123. doi:10.1002/1521-4095(20020816)14:16<1120::AID-ADMA1120>3.0.CO;2-9.
- ^ Venere, Emil (9 October 2014). "Electrically conductive plastics promising for batteries, solar cells". Purdue University News. Retrieved 13 July 2017.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}