비닐 양이온
Vinyl cation비닐 양이온은 알켄 카본에 양전하를 띠는 카보케이션입니다.그것의 경험식은 CH이다
2+
3.보다 일반적으로 비닐기 양이온은 양전하를 갖는 탄소가 이중결합의 일부이며 sp 하이브리드가 이루어지는 이치환된 3가 탄소이다.화학 문헌에서 치환된 바이닐 양이온은 종종 비닐 양이온으로 불리며 CH 변이체만
2+
3 지칭하는 것이 아니라 넓은 등급으로 지칭하는 것으로 이해된다.비닐 양이온은 4면 조정되지 않은 탄소 원자를 포함하는 반응성 중간체의 주요 유형 중 하나이며, 관찰된 다양한 반응성 경향을 설명하기 위해 필요합니다.예를 들어 강산에 의한 알킨 양성자화를 통해 알킨에 [3]대한 친전자성 첨가 중뿐만 아니라 용해 [1][2]반응에서 반응성 중간체로 비닐 양이온이 관찰된다.sp 혼성으로부터 예상대로, 비닐 양이온은 선형 형상을 선호합니다.비닐카티온과 관련된 화합물은 알릴카보카보레이션, 벤질카보레이션, 아릴카보레이션 등을 포함한다.

라디칼 및 카르보니온과 같은 다른 반응성 중간체와 비교했을 때, 비닐 양이온은 오랫동안 잘 이해되지[4] 않았고 처음에는 반응성 중간체로 형성하기에는 너무 높은 에너지로 간주되었다.비닐 양이온은 알킬 [5]아세테이트를 생성하는 알콕시아세틸렌의 산 촉매 가수분해에 대한 반응 중간체로 1944년에 처음 제안되었다.이들의 활성 수화 반응의 첫 단계인 속도 제한 단계에서 비닐 양이온 반응 중간체가 제안되었다. 양전하는 공식적으로 다이코랄 탄소에 존재하는 것으로 여겨졌다.문헌에서 이러한 전환 상태를 발견할 수 있는 것은 이번이 처음이다.
역사
Grob과 Cseh가 그들의 주요 [6]연구에서 알파-비닐 할로겐화물의 용해 반응에서 비닐 양이온 형성을 감지하면서, 15년이 지나서야 이 아이디어가 다시 검토되었다.사실, 이러한 공헌으로 Grob은 "비닐 양이온의 아버지"[7]로 불리고 있다.1960년대는 바이닐 양이온 관련 연구들이 쏟아져 나왔고, 동역학 자료들은 이 종의 존재에 대한 논쟁을 주도했다.예를 들어 노이스와 동료들은 페닐포로피올산의 [8]산촉매 수화에서 비닐 양이온의 형성을 보고했다.저자들은 속도 제한 단계에서 벤질 카본에서 큰 양의 전하가 발생하며, 이는 반응이 비닐 양이온 전이 상태를 통해 진행됨을 나타낸다.Noyce가 설명한 비닐 양이온의 접근성을 설명하기 위해 과접합 및 수소 결합을 유발하였다.
비닐 양이온 생성

비닐 양이온은 용해 반응 중에 반응성 중간체로 관찰되었습니다.S1N 화학과 일관되게, 이러한 반응은 1차 동력을 따른다.일반적으로 할로겐화비닐의 [10]존재 하에서 질산은 할로겐화비닐을 침전시키지 않으며, 이 사실은 역사적으로 양이온종의 [4]존재에 대한 논쟁을 위해 사용되었다.1970년대에 "슈퍼" 이탈 그룹의 도입으로 상당한 수명을 [11]가진 비닐 양이온 반응성 중간체를 생성할 수 있게 되었습니다.트리플루오로메탄술폰산염(트리플루오로메탄술폰산염) 및 비아플레이트(비아플루오로부탄술폰산염)와 같은 우수한 이탈기는 S1N 반응성에 매우 취약하다.이 슈퍼 탈퇴 그룹을 활용함으로써 연구진은 처음으로 비닐 양이온의 존재에 대한 추측을 뛰어넘을 수 있었다.

다른 이탈 그룹, 예를 들어 고가의 요오드 뮤티(고전적[13] 삼엽산염보다 100만 배 더 나은 이탈 그룹)도 그러한 목적을 위해 이용되었다.힝클과 동료들은 고가의 페닐리오도 전구체로부터 많은 알케닐(아릴) 요오드늄 삼산염을 합성했다.이 방법에서 E- 및 Z-비닐 삼산염은 헤테로 분해된 탄소-요오드 결합을 분해한 후 삼산염에 의해 양이온을 포착한 후에 형성된다.E- 및 Z-비닐 삼산염 제품의 존재는 1차 비닐 양이온 반응성 중간체의 형성을 지원하며, S2 화학을N 통해 둘 다 하나의 이성질체만 [9]형성된다.
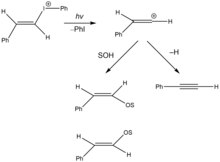
최근에는 광화학 용해 반응에서 비닐 양이온 반응성 중간체가 생성되고 있다.오른쪽 그림은 요오드화 비닐염의 광화학 용해를 이종분해 탄소-요오드 결합을 통해 분해하여 비닐 카보케이션과[14] 요오드벤젠을 생성한다.반응성 중간체는 용매에 의한 친핵성 공격을 받아 E 및 Z-에놀 에테르 이성질체를 생성하거나 베타 수소 제거를 일으키기 쉽다.
순환 비닐 양이온 생성
순환 비닐 양이온의 발생은 링 시스템의 크기에 따라 다르며, 작은 링에 있는 비닐 양이온은 생성하기가 더 어렵습니다.이러한 경향은 비닐 양이온이 선형 [15]배치를 선호한다는 것을 보여주는 계산에 의해 뒷받침된다.3원환계에서의 높은 변형률로 인해 가장 작은 고리형 비닐 양이온인 사이클로프롭-1-에닐 양이온의 생성은 여전히 어렵다.[16]다른 비닐 양이온을 생성하기 위해 사용되는N S1 용해 화학은 사이클로프롭-1-에닐 양이온에 대해 입증되지 않았습니다.이것은 아직 풀리지 않은 화학적인 도전이다.
비닐 양이온 구조
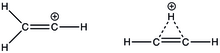
가장 단순한 비닐 양이온인
2+
3 CH는 두 가지 가능한 구조, 즉 고전적인 선형 구조 또는 비전통적인 브리지 구조를 가질 수 있습니다.ab initio 계산 결과 브리지 구조가 기존 구조보다 5.0 kcal/mol [17]더 안정적인 것으로 나타났습니다.그러나 동등한 알킬기를 가진 치환 비닐 양이온의 경우 선형 구조는 C와 [18]H NMR에 의해 지지되며, 비닐 양이온의 선형 구조의 첫 번째 실험 증거는 b-silyl 비닐 양이온의 X선 구조였다.다핵 NMR 분광법을 이용하여 화합물은 단일 Si NMR 신호를 보였으며, 이는 두 Si가 초공합을 통해 카보카피케이션과 동등하고 탈국소화됨을 의미한다.비닐 양이온은 C=C+ 스트레칭의 경우 1987cm의−1 강도 IR 피크를 가집니다.더 중요한 것은 비닐 양이온 탄소와 치환된 알킬의 첫 번째 탄소의 결합각을 약 [19]180으로o 측정했다는 것이다.
비닐 양이온 안정성
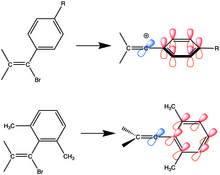
처음에는 비닐 양이온과 비닐 전구체 간의 에너지 차이가 크기 때문에 비닐 양이온의 존재 여부가 의문시되었다.일단 안정적인 비닐 양이온 중간체를 삼산염, 비아플레이트 등 양호한 이탈기를 가진 비닐 화합물의 용해로 얻을 수 있고 전자공여기에 의해 안정화 될 수 있다는 것이 확인되자 상당한 진전이 이루어졌으며 안정적인 비닐 양이온장이 생성되었다.
연구된 최초의 비닐 양이온 중 하나는 전자 기증 부분을 가진 아릴 치환기를 가지고 있었다.아릴비닐 화합물은 공진에 의해 안정화된다.이탈기를 제거할 때 빈 p-궤도는 페닐링의 공역계에 수직이기 때문에 비닐 빈 p-궤도가 페닐링의 p계와 동일 평면일 경우에만 전이 상태에서 공진 안정화를 달성할 수 있다.직교위치에 스테릭 부피를 더하면 페닐링이 비닐카본과 직교하지만 빈 p궤도와 동일평면을 이루므로 접합이 향상된다.


아릴비닐 양이온과 마찬가지로 디에닐 및 알렌 양이온도 결합에 의해 안정화된다.다시 말하지만, 공진 안정화를 달성하기 위해서는 공진계의 이중 결합이 빈 p-궤도와 동일 평면이어야 합니다.알렌 양이온에서는 정전하가 구조 전체에 걸쳐 잘 분포되어 있다.
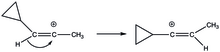
사이클로프로필비닐 양이온은 안정화에 대한 비전통적 접근을 보인다.이등분된 구조일 때, 빈 p-궤도와 사이클로프로필 고리 사이에 적절한 중첩이 있어 안정화가 달성된다.다른 형태인 수직 구조에서 빈 p-궤도는 링 시스템에 수직이다.사이클로프로필 고리의 안정화 힘은 매우 커서 (E)- 및 (Z)-3-시클로프로필-2-프로페닐 삼산염 용해와 [20]같은 배열에서 구동 열역학적 힘이 되었다.
비닐 양이온 안정성에 대한 치환 효과
| 치환기 | 안정화# | α 치환기의 전자 효과 | ||
| 유도^ | - - donationdonation | 초결합 | ||
| -CH2 | + | - | +* | |
| -CH3 | + | + | ||
| - Cl. | + | - | +* | |
| -Br | + | - | +* | |
| - 나 | + | - | +* | |
| -F | - | -* | + | |
| -NH2 | + | - | +* | |
| - 오 | + | - | +* | |
| - SH | + | - | +* | |
| -C6H5 | + | +* | ||
| -CF3 | - | - | ||
| - CHF2 | - | - | ||
| -아니요2 | - | - | ||
| -CnN | + | - | +* | |
| - CHY2*** | + | - | +* | |
| - Si(CH3)3 | + | + | ||
| -C(O)H | - | +/-** | ||
| -쿠우 | - | +/-** | ||
| -C(CH3)2OH | + | - | ||
| -CchCH | + | - | +* | |
표 1: α위치에서의 비닐 양이온 안정화를 담당하는 전자효과
^ '-' 전자전달, '+' 전자전달
# '+'는 안정화를 나타내고 '-'는 중성 알켄 당량에 대한 치환 비닐 양이온의 불안정화를 나타낸다.
*2개 이상의 전자적 효과를 보이는 대체물질에 대해 (탈)유효성을 일으키는 가장 강력한 인자를 나타낸다.
** 치환기는 카보닐 카본에서 유도적으로 인출되며 카보닐 산소로부터 작은 전자 이탈을 보입니다.
*** Y = -F, -Cl, -Br, -I, -OH, -CN, -CF3
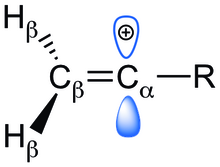
p-결합에 수직인 빈 p-궤도가 있으면 비닐 양이온에 원치 않는 불안정성이 발생합니다.이러한 내재적 불안정성은 카보케이션에서 전하를 감소시키는 a-치환 성분과의 바람직한 상호작용을 통해 감소될 수 있다.ab initio 계산법은 구조 내의 엔탈피, 결합 길이, 결합 순서 및 전하의 변화를 감시함으로써 치환기의 안정화 또는 불안정화 효과를 나타내기 위해 사용되어 왔다.

치환기가 비닐 양이온의 안정성에 영향을 미칠 수 있는 세 가지 전기적 효과가 있을 수 있습니다.탄소로부터 더 많은 전자 밀도를 끌어냄으로써 양이온을 불안정하게 하거나 더 많은 전자 밀도를 기여함으로써 안정화시킬 수 있습니다.카르보케이션 양전하는 불포화탄소계 또는 헤테로원자 치환기에 의해 p-기증 및/또는 메틸렌/메틸 치환기에 의한 C-H 과접합을 통해 완화될 수 있다.또한 유도 효과는 치환기가 전자공여인지 또는 인출인지에 따라 안정되거나 불안정해질 수 있습니다.이 세 가지가 모두 캐티온의 전반적인 안정성에 영향을 미치기 때문에 개별 전자 효과는 다른 효과와 분리할 수 없습니다.
비닐 양이온의 경우 중성 알켄 유사체와 상대적인 안정성을 비교할 수 있습니다.a-치환물의 안정화 특성을 얻기 위해 이등변성 반응을 이용하여 반응 엔탈피를 얻어 치환된 비닐 양이온과 중성 알켄 전구체 간의 엔탈피 차이를 계산하였다.이 방법은 실험적으로 결정된 열화학적 값에 대해 벤치마킹할 수 있기 때문에 유리하다.계산은 비닐 양이온의 브리지된 비고전적 구조에서 초기화됩니다. 이는 전지구적 최소값이기 때문입니다.
초기 연구에서는 비닐 양이온 안정성에 대한 전자적 영향을 조사하기 위해 4가지 치환기(-CH=CH2, -F, -Cl, -CH3)를 연구하였다.서로 다른 a-치환성분은 중성 알켄 성분과 비교하여 비닐 양이온의 구조적 변화를 유도한다.이러한 변화는 존재하는 전자적 효과에 기인할 수 있다.비닐 양이온에서는 C-R 및 Cb=C 결합길이가 현저하게 감소하여 C-R 및 C=Ca 사이의a 전자공여 또는 유도를 나타낸다.한편, C-Hb 결합 길이의 증가는 양이온의 열역학적 안정성과 반비례하는 강한 초공역적 효과를 의미한다.C에서a C-H 결합과 빈 p-궤도의 중첩이 양호하기 때문에 안정화가 가능하다.인접한 C-Hb 결합과 –CH3 치환기 때문에 모든 구조에서 과접합이 뚜렷하다.
등각 반응에서 얻은 엔탈피 계산은 상당히 정확하고 실험 데이터와 좋은 상관관계를 보여준다.스태빌라이제이션은 -F < -Cl3 < -CH < -CH2 = CH ) 。비닐 양이온을 7kcal/mole만큼 불안정하게 만드는 불소를 제외한 모든 치환기는 안정성을 제공합니다.이러한 현상은 비닐과 에틸 양이온에 대한 a-불소 치환기 효과를 비교함으로써 설명할 수 있다.에틸 양이온에서는 불소가 카보카치온을 안정화시킵니다.비닐과 에틸카티온의 불소 안정화 능력이 극명한 차이를 보이는 것은 a-카본의 교배 차이 때문이다.비닐 양이온은 전기음성 sp-하이브리드화 탄소를 더 많이 가지고 있기 때문에 유도 효과가 더 두드러질 것입니다.전기음성 sp-하이브리드화 탄소가 불소와 상호작용하는 것은 구조를 크게 불안정하게 한다.이러한3 현상은 -CH=CH2 치환기와 -CH=CH2 치환기를 비교할 때도 덜 나타난다.
불소 및 염소와 같은 헤테로아토늄은 높은 전기음성도와 p-전자로 인해 유도성(전자 인출) 및 p-기증성 전자 효과를 모두 나타낼 수 있습니다.안정화는 두 전자 효과 사이의 균형에 따라 달라집니다.불소의 경우 유도를 통한 불안정화가 지배적이며 공명은 상당히 약하다.염소의 경우, 공명은 유도에 대항하기에 충분하므로 전체적으로 효과가 안정됩니다.
유도 인출/기증 및 p-기증 치환기의 경우 일부 부분전하가 R군과 C군에 존재한다a.그러나 네 가지 치환기에 대한 R과a C의 전하 크기 추세는 반비례 관계가 있다.또한 C=C와a C-R의a 결합b 차수가 증가하며, 이는 대응하는 결합 길이 변화와 일치한다.
치환기의 작은 표본 크기에서는 R에 대한 결합 차수 증가와 전하 분포, 치환기에 의한 안정화 사이의 상관관계가 관찰되지 않았다.그러나 안정화는 C-Hb 결합 연장과 상관관계를 보였다.
상기의 메커니즘에 근거해, 광범위한 비닐 양이온 a-하위 성분은, 전자 효과에 의해서 분류할 수 있고, 안정성의 정도는, 이러한 효과 사이의 미묘한 균형에 의해서 좌우된다.
-NH2, -OH, -SH와 같은 단일 쌍 함유 치환기는 p-기부가 유도 인출 효과를 극복하여 안정화되고 있다.-CH=CH2, -CH와65 같은 복합계는 강한 p-성분 때문에 안정되고 있다.-CF3 및 –NO와2 같은 매우 불안정한 치환기는 유도성 전자 이탈만 보입니다.-CN과 같이 약하게 불안정한 치환기는 약한 p-기증 효과를 가지며, 전자 인출에 의한 유도를 완전히 억제하지 않는다.
다른 전자 효과가 방해가 되기 때문에 이원자 a-치환 성분의 유도 효과를 분리하는 것은 전적으로 타당하지 않다.그러나 기능성 그룹의 유도 효과를 조사할 수 있는 한 가지 방법은 헤테로아톰이 비닐 양이온(-CHY2)에서 떨어진 메틸렌기일 수 있는 b-치환 효과를 조사하는 것이다.매우 작거나 p 기증을 보이지 않는 CHY2 그룹에서 치환기의 –CH-2 그룹에서 과공성 효과의 차이는 매우 작다.따라서 전체적인 안정성은 이제 유도력에 의해서만 구동되는 b-치환 효과와 상관될 수 있다.순수하게 유도적으로 기능하는 그룹의 능력만을 비교하면 CN > CF3 > F > Cl > Br > OH 순이며, 메틸기와 동등한 불안정 에너지도 있습니다.
대부분의 경우 치환기는 둘 이상의 전자 안정화 효과를 보입니다.통상, 복수의 결합에 의해서 발생하는 헤테로 원자와의 유도 효과는, 같은 헤테로 원자로부터의 p기증함으로써 상쇄할 수 있다.예를 들어 절대 b유도력에 근거해 -CN은 CF보다3 유도성이 높지만 CN의 질소로부터 p기증이 있을 수 있기 때문에 유도성이 저하된다.F, Cl, Br, OH와 같은 일반적인 헤테로 원자 치환기에서 안정성은 높은 전자 인출 능력과 함께 감소한다.그러나, p기증은 여전히 C-R 결합 감소로 인해 일어나는 것으로 여겨진다.
카르보닐 치환기는 주로 비닐 양이온 옆에 고도로 양의 카르보닐 카본이 있고 p 공여가 없기 때문에 불안정하다.
비닐 양이온과 에틸 양이온의 치환 효과를 비교하여 안정화에 의한 교배 효과를 조사하는 것이 유용하다.일반적으로 비닐 양이온은 에틸 양이온과 비교하여 치환기에 의해 안정성이 더 높아지는데, 이는 비닐 양이온이 원래 안정성이 떨어지기 때문입니다.-F, -OH, -NH와2 같은 강한 유도성 전자 인출기는 에틸카티온스페2 하이브리드에 비해 비닐카티온스페 하이브리드가 전기음성성이 높기 때문에 에틸카티온스페 하이브리드에 비해 유도성 불안정성이 보다 뚜렷하다.반면 α-Si(CH3)3 치환기의 경우 p-전자가 없기 때문에 비닐 양이온에 대한 안정성이 높다.
결합순서 측면에서 치환기를 안정화하면 C-R, Cα=Cβ, C-Hβ 결합순서가 증가한다.-CF3, -CHF2 및 -CHX에서2 결합 순서의 작은 증가가 관찰되며, -CH=CH2, -I 또는 -SH와 같이 p 또는 p 전자를 기증할 수 있는 치환기에서 결합 순서의 큰 증가가 관찰된다.
화학반응 중 비닐 양이온 중간체
친전자성 첨가물
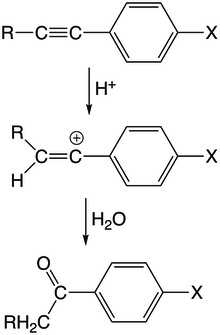
친전자성 부분이 불포화탄소를 공격할 때 비닐 양이온 중간체를 형성할 수 있다.이것은 전자 친필이 알킨이나 알렌과 반응할 때 달성될 수 있다.이러한 반응에서, 양전자 친필은 비닐 양이온을 형성하는 불포화 탄소 중 하나를 공격하고, 이 탄소는 이후 최종 생성물을 형성하기 위한 추가 반응 단계를 거친다.
아릴아세틸렌 유도체의 산촉매 수화에서 양성자가 최초로 삼중결합을 공격하여 아릴 치환탄소에서의 비닐카티온을 형성한다.중간은 비닐 양이온의 빈 p-궤도와 켤레 아릴 오비탈의 직교성 때문에 공명 안정화를 거의 경험하지 못한다.반응은 아세틸렌과 양성자 모두와 관련하여 첫 번째 순서이며 속도 결정 단계로서 아세틸렌의 양성자와 함께 이루어진다.단일 치환 아릴/알콕시 아세틸렌은 메틸 치환 당량에 비해 산성 수화에서 더 빠른 동력을 보인다.아릴아세틸렌에서 메틸기는 C-H 과접합으로 인해 수소에 비해 덜 안정화에 기여하는 것으로 나타나 알킬 양이온에서 관찰되는 안정화 추세를 역전시킨다.C-H 결합은 빈 p-궤도와 유의하게 중복될 수 있기 때문에 C-H 과접합은 중요한 요인이다.또 다른 가능한 설명은 수소 치환기의 크기가 작을수록 용해가 더 쉽게 일어나 더 큰 안정화에 기여할 수 있다는 것이다.
양성자 외에도, 다른 친전자 집단은 아세틸렌 부분을 공격할 수 있다.카르본산의 공격을 받으면 시스/트랜스 알켄 부가물이 형성될 수 있다.초기 양성자화 단계를 가진 할로겐화 수소와의 반응은 할로 치환 알케인을 형성한다.마지막으로 아다만틸케톤은 아세틸렌에 대한 아다만틸카티온 공격과 그에 따른 [24]수화작용에 의해 형성될 수 있다.
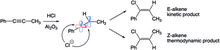
페닐프로펜의 하이드로겐화에서는 열역학적 및 운동학적 효과에 의해 두 가지 다른 알켄 생성물이 형성된다.선형 SP 하이브리드 비닐 양이온은 두 가지 다른 방향에서 할로겐의 공격을 받을 수 있습니다.덜 입체적으로 저해되는 쪽(수소)에서 공격받으면 E-알켄이 생성되고, 다른 쪽으로 공격받으면 Z-알켄이 형성된다.단시간 스케일에서는 부피가 작은 쪽의 공격이 선호되기 때문에 E-알켄이 바람직하지만, 장기간에 걸쳐서는 보다 안정된 Z-알켄(대측의 메틸기 및 페닐기)이 바람직하다.E-알켄은 초기에 형성되지만 양성자화 및 C-C 회전 [25]단계로부터 유래한 카보카케이션 중간을 통해 Z-알켄으로 이성화된다.

알킨을 둘러싼 인접 그룹은 분자 내 상호작용과 같은 비고전적 접근법을 통해 중간과 상호작용함으로써 반응 동력을 향상시킬 수 있다.3차 알코올에 인접한 알킨은 수산기의 산소가 2개의 탄소를 2개의 결합에 걸쳐 브리지하는 4원짜리 고리형 비닐 양이온 중간체이다.마찬가지로 5-클로로 치환된 1-펜틴에서 5원클로로늄환 중간체를 형성한다.중간체가 C-Cl5 위치에서 [24]헤테로 분해되기 때문에 비정상적으로 시프트된 생성물이 형성된다.
알렌의 친전자성 공격에서는 중심 카본에서 말단 부가물과 비닐 양이온을 형성하는 것을 선호하는 방식으로 이루어진다.알렌 그룹의 분극은 말단 탄소가 더 높은 전자 밀도를 가지고 핵 친균 공격을 받는 경향을 가지고 있다는 것을 보여준다.단, 말단부가 치환기에 의해 안정화되면 전자친이 중앙카본에 공격을 가함에 따라 알릴 형태의 양이온이 형성될 수 있다.비닐 양이온에 인접한 페닐링과 마찬가지로 완전한 공진 [24]안정화를 위해서는 결합 회전이 있어야 합니다.
비닐 양이온 재배열

반응 중에 형성되는 비닐 양이온 중간체는 전위되는 경향이 있다.이러한 재배열은 크게 두 가지 클래스로 분류할 수 있습니다. 이중 결합으로의 이행과 이중 결합을 통한 재배열입니다.첫 번째 범주는 알릴 양이온 형성을 유도하는 1,2-시프트를 포함하고, 두 번째 범주는 또 다른 비닐 양이온 이성질체를 형성한다.
비닐 양이온은 1,2-수소 시프트를 거쳐 알릴 안정화 양이온을 형성한다. 1,2-수소 시프트는 알킬 양이온에서 상당히 일반적이며 NMR 시간 척도에서 빠르다.그러나 비닐 양이온에서는 배열 생성물이 열역학적으로 안정되어 있어도 이러한 배열은 드물다.아릴 치환 비닐 양이온과 마찬가지로 선형 비닐 양이온에서 비선형 알릴 양이온으로의 변환 시 상호작용하는 오비탈은 직교하며 비평면 전이 상태를 통과하기 때문에 재배치가 어렵다.이는 알킬 양이온에 비해 비닐 양이온에서 1,2-수소화물 이동의 높은 활성화 에너지에서 명백하다.이러한 현상이 관찰되는 반응의 예로는 디알킬 치환 알킨의 양성자화 및 트리플루오로에탄올의 이스프로필비닐 트리플루로메탄올산염의 용해 등이 있다.

비닐 양이온에서도 1,2 메틸 시프트가 발생하며, 1,2-수소 시프트와 마찬가지로 알킬 양이온 당량에 비해 활성화 장벽이 높다.알킨의 양성자화에서 1,2-수소화 및 1,2-메틸 이동이 모두 발생할 수 있으며, 그 선호도는 알킬 치환기에 따라 결정되며, 이는 결과 알릴 양이온 생성물을 지시하기 때문이다.t-부틸 치환기는 1,2-메틸 시프트가 바람직하며, 이소프로필 치환기는 1,2-수소 시프트가 대신 발생한다.고리형 알켄도 용해 시 1,2-메틸 변화를 보인다.

스피로-비닐 삼산염의 용해에서 합치된 과정을 통한 비닐 양이온 중간체의 형성은 완전히 다른 고리 구조의 형성을 수반하는 전위를 더욱 촉진한다.비닐 양이온의 재배치를 통해서도 링의 팽창을 실현할 수 있다.
재배열의 두 번째 클래스는 비닐 양이온 이성질체를 형성하기 위해 재배열합니다.이 과정은 용매, 친핵물질의 성질, 화합물 내 부분에 크게 의존한다.1차 비닐 양이온에서는 수소의 전자공여 능력이 낮기 때문에 1차 비닐 양이온의 안정성이 낮기 때문에 1,2-수소화물이 발생할 가능성이 낮다.그러나, 이것은 1-메틸-2-페닐비닐 삼산염과 같이 생성된 비닐 양이온이 공진 안정화되는 특수한 경우에 여전히 관찰된다.

but-2-yne에 tert-부틸 양이온을 첨가하면 메틸 변화가 관찰된다.형성되는 펜타알릴 양이온은 단일 1,3-메틸 시프트 또는 두 개의 연속적인 1,2-메틸 시프트의 결과일 수 있습니다.이중 결합을 통한 재배열은 순환 시스템의 크기를 변경할 수도 있습니다.메틸 치환 시클로헥세닐 트리페이트의 용해에서는 직선 구조로 인해 전위 생성물보다 선호도가 낮아 전위 생성물과 비전위 생성물이 거의 동일한 양으로 형성된다.그러나 메틸에네시클로펜탄 전위 생성물에 약간의 변형이 있다는 점에 유의해야 한다.
마지막으로 할로겐은 비닐 양이온 시스템으로 이동하여 안정화시킬 수 있습니다.5-클로로펜틴과 트리플루오로아세트산의 반응에서 4개의 탄소에 걸쳐 교량 고리 구조를 형성하는 염소의 양성화와 1,4-시프트가 동시에 일어난다.이어서 트리플루오로아세트산이 말단에서 중간체를 공격하여 2-클로로펜트-4-에닐트리플루오로아세트산염을 형성한다.이 현상은 다른 할로겐에서도 관찰된다.예를 들어 플루오로알킨은 2개의 [18]부가물을 가진 제품을 형성할 수 있다.
순환 반응 중 비닐 양이온

케텐과 알렌은 서로 직교하는 파이 오비탈을 가지고 있기 때문에 열조건 하에서 [2+2]의 사이클로드 상태를 조화롭게 거친다.비닐 양이온 중간체는 2p의 오비탈을 가지며 동시에 친이성 오비탈과 겹칠 수 있기 때문에 동일한 과정을 거친다.2-부틴과 Cl의2 Smirnov-Zamkow 반응에서 시클로디션이 디클로로시클로부탄을 형성한다.알렌이 HCl과 반응할 때도 비슷한 반응이 관찰된다.시클로디션 후 양이온성 순환 중간체를 형성한 후 친핵자에 의해 공격을 받아 최종 [26]생성물을 형성한다.
할로겐화 비닐 양이온
할로겐화 반응용 말단 알킨에 할로겐화물(H-X) 화합물을 첨가하여 비닐 양이온 중간체를 형성하는지 여부에 대한 논란이 있다.대안으로, 일부에서는 이 경우에 H와 Br의 추가가 실제로 [citation needed]일치한다고 믿는다.
레퍼런스
- ^ Okuyama, T. (2002). "Solvolysis of Vinyl Iodonium Salts. New Insights into Vinyl Cation Intermediates". Acc. Chem. Res. 35 (1): 12. doi:10.1021/ar0100374.
- ^ Gronheid, R (2001). "Thermal and Photochemical Solvolysis of (E)- and (Z)-2-Phenyl-1-Propenyl(phenyl)iodonium Tetrafluoroborate: Benzenium and Primary Vinylic Cation Intermediates". J. Am. Chem. Soc. 123 (36): 8760. doi:10.1021/ja010861n.
- ^ Walkinshaw, Andrew J.; Xu, Wenshu; Suero, Marcos G.; Gaunt, Matthew J. (2013). "Copper-Catalyzed Carboarylation of Alkynes via Vinyl Cations". Journal of the American Chemical Society. 135 (34): 12532–12535. doi:10.1021/ja405972h. PMID 23947578.
- ^ a b Stang, P.J. (1979). Vinyl Cations. New York: Academic Press. p. 2.
- ^ Jacobs, Thomas L.; Searles, Scott (1944-05-01). "Acetylenic Ethers. IV.1 Hydration". Journal of the American Chemical Society. 66 (5): 686–689. doi:10.1021/ja01233a007. ISSN 0002-7863.
- ^ Grob, C.A. (1964). "Die Solvoltische Decarboxylierung von α,β-Ungesättigeten β-Halogensäuren Fragmentierungsreaktionen, 9. Miteilung". Helv. Chim. Acta. 47 (6): 1590. doi:10.1002/hlca.19640470621.
- ^ Miyamoto, K. (2009). "Facile Generation of a Strained Cyclic Vinyl Cation by Thermal Solvolysis of Cyclopent-1-Enyl-λ3-Bromanes". Angew. Chem. Int. Ed. 48 (47): 8931–4. doi:10.1002/anie.200903368. PMID 19830754.
- ^ Noyce, D. (1965). "Concerning the Acid-Catalyzed Hydration of Acetylenes". J. Am. Chem. Soc. 87 (10): 2295. doi:10.1021/ja01088a042.
- ^ a b Hinkle, R.J. (1999). "Primary Vinyl Cations in Solution: Kinetics and Products of a,a-Disubstituted Alkenyl(aryl)iodonium Triflate Fragmentations". J. Am. Chem. Soc. 121 (32): 7437–7438. doi:10.1021/ja9916310.
- ^ Shriner, R.L. (1964). Systematic Identification of Organic Compounds. New York: Wiley.
- ^ Hanack, Michael (1970-07-01). "Vinyl cations in solvolysis reactions". Accounts of Chemical Research. 3 (7): 209–216. doi:10.1021/ar50031a001. ISSN 0001-4842.
- ^ Stang, P.J. (1979). Vinyl Cations. New York: Academic Press. p. 213.
- ^ Okuyama, Tadashi; Takino, Tomoki; Sueda, Takuya; Ochiai, Masahito (1995-03-01). "Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion-Molecule Pair". Journal of the American Chemical Society. 117 (12): 3360–3367. doi:10.1021/ja00117a006. ISSN 0002-7863.
- ^ a b Tidwell, Thomas T.; P.), Richard, J. P. (John (2003-01-01). Advances in physical organic chemistry. Vol. 37. Academic. ISBN 978-0120335374. OCLC 51840423.
- ^ Mayr, Herbert; Schneider, Reinhard; Wilhelm, Dieter; Schleyer, Paul V. R. (1981-12-01). "Vinyl cations. Comparison of gas-phase thermodynamic and solvolysis data with ab initio MO calculations" (PDF). The Journal of Organic Chemistry. 46 (26): 5336–5340. doi:10.1021/jo00339a015. ISSN 0022-3263.
- ^ Grob, C. A.; Csapilla, J.; Cseh, G. (1964-01-01). "Die solvoltische Decarboxylierung von α,β-ungesättigeten β-Halogensäuren Fragmentierungsreaktionen, 9. Miteilung". Helvetica Chimica Acta. 47 (6): 1590–1602. doi:10.1002/hlca.19640470621. ISSN 1522-2675.
- ^ a b c Pople, J.A. (1987). "The structure of the vinyl cation". Chemical Physics Letters. 137 (1): 10–12. Bibcode:1987CPL...137...10P. doi:10.1016/0009-2614(87)80294-4.
- ^ a b c d e f g h i j k l Shchegolev, A A; Kanishchev, M I (1981). "Rearrangements in Vinyl Cations". Russian Chemical Reviews. 50 (6): 553–564. Bibcode:1981RuCRv..50..553S. doi:10.1070/rc1981v050n06abeh002650.
- ^ Müller, Thomas; Juhasz, Mark; Reed, Christopher A. (2004-03-12). "The X-ray Structure of a Vinyl Cation". Angewandte Chemie International Edition. 43 (12): 1543–1546. doi:10.1002/anie.200352986. ISSN 1521-3773. PMID 15022228.
- ^ a b c d e Hanack, Michael (1976-10-01). "Stabilized vinyl cations". Accounts of Chemical Research. 9 (10): 364–371. doi:10.1021/ar50106a004. ISSN 0001-4842.
- ^ van Alem, Kaj; Lodder, Gerrit; Zuilhof, Han (2000-03-01). "α-Substituted Vinyl Cations: Stabilities and Electronic Properties". The Journal of Physical Chemistry A. 104 (12): 2780–2787. Bibcode:2000JPCA..104.2780V. doi:10.1021/jp9935743. ISSN 1089-5639.
- ^ van Alem, Kaj; Lodder, Gerrit; Zuilhof, Han (2002-11-01). "Delocalization Does Not Always Stabilize: A Quantum Chemical Analysis of α-Substituent Effects on 54 Alkyl and Vinyl Cations". The Journal of Physical Chemistry A. 106 (44): 10681–10690. Bibcode:2002JPCA..10610681V. doi:10.1021/jp021766j. ISSN 1089-5639.
- ^ a b c Advances in Physical Organic Chemistry. Academic Press. 1971-12-31. p. 185. ISBN 9780080581484.
vinyl cation advances in physical organic chemistry modena.
- ^ a b c Modena, Giorgio (1971). "Vinyl cations". Advances in Physical Organic Chemistry. 9: 185–280.
- ^ a b Organic Chemistry (Second ed.). Oxford, New York: Oxford University Press. 2012-05-04. ISBN 9780199270293.
- ^ a b Fleming, Ian (2010). Molecular Orbitals and Organic Chemical Reactions, Reference Edition - Fleming - Wiley Online Library. doi:10.1002/9780470689493. ISBN 9780470689493.


