에나민
Enamine
에나민은 알데히드나 케톤이 2차 아민으로 응결하여 얻은 불포화 화합물이다.[1][2] 에나민은 다재다능한 매개체다.[3][4]
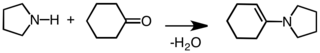 에나민을 주기 위한 응결.[5]
에나민을 주기 위한 응결.[5]

"에나민"이라는 단어는 알켄의 접미사로 사용되는 아픽스 en-과 뿌리 아민에서 유래되었다. 이는 알켄(엔-)과 알코올(-oll)을 모두 포함하는 기능군인 에놀과 비교할 수 있다. 에나민은 에놀의 질소 아날로그로 간주된다.[6]
만약 질소 대체물 중 하나가 수소 원자 H라면, 그것은 이미인의 태토적인 형태다. 이것은 보통 이미인으로 재배열된다. 그러나 몇 가지 예외(예: 아닐린)가 있다. 에나민-이미네 토토머리즘은 케토-에놀 토토머리즘과 유사한 것으로 간주될 수 있다. 두 경우 모두 수소 원자는 이질 원자(산소 또는 질소)와 두 번째 탄소 원자 사이에서 위치를 전환한다.
에나민은 좋은 핵성분과 좋은 염기 둘 다이다. 탄소 기반 핵물질로서의 그들의 행동은 다음과 같은 공명 구조와 관련하여 설명된다.

포메이션
에나민은 미약하기 때문에 화학적으로 유용한 모이티로 시중에서 구할 수 있는 시약으로부터 쉽게 생산될 수 있다. 에나민 생산을 위한 일반적인 경로는 2차 아민이 함유된 α-수소를 함유한 케톤(Stork, 1963) 또는 알데히드(Mannich/Davidsen, 1936)종의 산성 촉매 핵포필 반응을 통해 이루어진다. 반응 아민의 pKa가 충분히 높은 경우(예: pKa가 11.26인 피롤리딘) 산성 촉매제가 항상 필요한 것은 아니다. 그러나 반응 아민의 pKa가 낮을 경우 첨가 단계와 탈수[7] 단계(일반 탈수제에는 MgSO와4 NaSO가24 포함된다)[8]를 통해 산성 촉매제가 필요하다. 일차 아민은 보다 열역학적으로 안정된 이미인 종의 우선적 형성으로 인해 보통 에나민 합성에 사용되지 않는다.[9] 메틸케톤 자가 응축은 부작용 혼합물에 TiCl을4[10] 첨가함으로써 피할 수 있는 부작용이다(물청소기 역할을 하기 위해).[11][12] 알데히드가 2차 아민과 반응하여 카르비놀라민 중간을 통해 에나민을 형성하는 예는 다음과 같다.

반응
알킬화
에나민은 에놀에 비해 핵소독성이 강하지만 선택적으로 반응할 수 있어 알킬화 반응에 유용하다. 에나민 뉴클레오필드는 할로윈에탄스를 공격하여 알킬화 이미늄 소금 중간을 형성할 수 있으며, 이 중간은 가수분해하여 케톤(에나민 합성의 시작 물질)을 재생시킬 수산화 아이미늄 소금 중간은 에나민 합성의 시작 물질이다. 이 반응은 길버트 황새에 의해 개척되었고, 때때로 그것의 발명가의 이름으로 언급되기도 한다. 이와 유사하게, 이 반응은 효과적인 아세틸화 수단으로 사용될 수 있다. 이 반응에는 벤질릭, 아군 할레이드를 포함한 다양한 알킬링 및 아틸링제를 사용할 수 있다.[13]

아킬레이션
에나민 알킬화와 매우 유사한 반응으로 에나민은 아틸화 되어 최종 디카르보닐 제품을 형성할 수 있다. 에나민 시작 물질은 이미늄 소금 중간을 형성하는 아킬 할로겐에 뉴클레오필릭을 첨가하여 산에서 가수 분해할 수 있다.[14]

메탈에에에아민스
LiNR2와 같은 강력한 베이스는 이미인을 감응시키고 야금성을 형성하는데 사용될 수 있다. 메탈로네아민은 그들의 핵소독성 때문에 종합적으로 유용하다는 것을 증명할 수 있다. (그것들은 에놀레이트보다 핵소독성이 더 많다. 따라서 그들은 약한 전기영동체와 더 잘 반응할 수 있다(예를 들어, 그것들은 에폭시드를 여는 데 사용될 수 있다).[15] 가장 두드러지게, 이러한 반응은 키랄 중간 야금류로의 변환을 통해 케톤들의 비대칭 알킬화를 허용했다.[16]
할로겐화
β-산화임모늄 화합물은 디에틸에테르 용매에 할로겐화물이 함유된 에나민 반응을 통해 합성할 수 있다. 가수 분해는 α-할로 케톤을 형성하게 된다.[17] 염소화, 브로민화, 요오드화까지 가능한 것으로 나타났다. 일반적인 반응은 다음과 같다.

산화 커플링
에나민은 질산 암모늄과의 처리를 통해 에놀 실란과 효율적으로 교차 결합될 수 있다. 이러한 반응은 1935년에 나라사카 그룹에 의해 보고되었으며, 1,4 디케톤(모폴린 아민 시약에서 파생된)의 한 인스턴스뿐만 아니라 안정적인 에나민으로 가는 경로를 제공했다.[18] 후에, 이러한 결과는 나라사카 기판을 사용하여 1,4개의 디카르보닐을 좋은 수율과 함께 반감적으로 생산하기 위해 사용한 유기촉매의 개발로 맥밀런 그룹에 의해 이용되었다.[19] 아민이 존재하는 알데히드의 산화적 조광화는 에나민 형성에 이어 최종 피롤 형성을 통해 진행된다.[20] 대칭 피롤 합성을 위한 이 방법은 2010년 지아 그룹에 의해 피롤 함유 천연물의 합성을 위한 귀중한 새로운 경로로 개발되었다.[21]
애널링
에나민 화학은 로빈슨 경매의 1배트짜리 항억제적 버전을 생산하기 위한 목적으로 시행되었다. 로버트 로빈슨(Robert Robinson)이 1935년에 발표한 로빈슨(Robinson annation)은 케톤(ketone)과 메틸(methyl)비닐케톤(MVK)을 결합하여 시클로헥세논(Cylohexenone) 융합 링 시스템을 형성하는 염기성 반응이다. 이 반응은 좋은 입체감을 허용하는 치랄 에나민 중간체를 통해 진행되도록 프롤라인에 의해 촉매될 수 있다.[22] 이것은 특히 자연 제품 합성 분야에서 특히 중요하다. 예를 들어, 보다 복잡한 생물학적으로 활성 분자의 필수적인 구성 요소인 Wieland-Mescher ketone의 합성을 위해서 말이다.[23][24]
반응도
에나민은 에나민보다 반응성에 대한 산/베이스 활성화가 덜 필요한 핵물질로 작용한다. 그들은 또한 더 적은 부작용과 더 큰 선택성을 제공하는 것으로 나타났다. 다른 에나민 종류들 사이에 반응성의 경사가 있으며, 케톤 에나민이 알데히드보다 더 큰 반응성을 제공한다.[25] 순환 케톤 에나민은 5개의 막이 달린 링이 질소에서의 평면적 순응으로 인해 가장 반응성이 높은 반응도 추세를 따르며, 트렌드 5>8>6>7 (7개의 막이 달린 링은 가장 반응성이 낮다)을 따른다. 이러한 경향은 질소 단독 쌍 궤도 상에 p-문자의 양 - p-Orbital이 알켄 π-궤도에 기증을 허용하기 때문에 더 큰 핵소독성에 해당하는 p-문자의 양에 기인한다. 유사하게, N 단독 쌍이 아민 해이에서 스테레오 전자 상호작용에 참여한다면, 단독 쌍은 비행기에서 튀어나와 (피라미드화 될 것이다) 인접한 π C-C 결합으로 기부를 위태롭게 할 것이다.[26] [27]

온도 변화, 용매 변화, 기타 시약의 양 및 전기영양 유형을 포함하여 질소 센터의 스테릭/전자장치를 변경하는 것 외에 에나민 반응성을 조절하는 많은 방법이 있다. 이러한 파라미터를 튜닝하면 E/Z 에나민의 우선 형성이 가능하며 케톤 시작 물질에서 더/더 적은 대체 에나민의 형성에 영향을 미친다.[28]
참고 항목
- 엔더스 SAMP/RAMP 히드라존-알킬화 반응
- 하조스-파리시-에더-사우어-위처트 반응
- 마이클 엑스트라
- 네니체스쿠 인도레 합성
- 조직투석
- 로빈슨 애닝
- 황새 에나민 알킬화
- 소르페 반응
- 플루옥시메스테론
참조
- ^ Clayden, Jonathan (2001). Organic chemistry. Oxford, Oxfordshire: Oxford University Press. ISBN 978-0-19-850346-0.
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ 에나민: 합성: Structure, and Reactions, Second Edition, Gilbert Cook (편집자) 1988년, 뉴욕주 마르셀 데커. ISBN 0-8247-7764-6
- ^ R. B. Woodward, I. J. Pachter, and M. L. Scheinbaum (1974). "2,2- (Trimethylenedithio)cyclohexanone". Organic Syntheses. 54: 39.
{{cite journal}}: CS1 maint : 복수이름 : 저자목록(링크); - ^ R. D. Burpitt and J. G. Thweatt (1968). "Cyclodecanone". Organic Syntheses. 48: 56.; Collective Volume, vol. 5, p. 277
- ^ Imines and Enamines PharmaXChange.info
- ^ Capon, Brian; Wu, Zhen Ping (April 1990). "Comparison of the tautomerization and hydrolysis of some secondary and tertiary enamines". The Journal of Organic Chemistry. 55 (8): 2317–2324. doi:10.1021/jo00295a017.
- ^ Lockner, James. "Stoichiometric Enamine Chemistry" (PDF). Baran Group, The Scripps Research Institute. Retrieved 26 November 2014.
- ^ Farmer, Steven (2013-10-16). "Enamine Reactions". UC Davis Chem Wiki.
- ^ Carlson, R; Nilsson, A (1984). "Improved Titanium Tetrachloride Procedure for Enamine Synthesis". Acta Chemica Scandinavica. 38B: 49–53. doi:10.3891/acta.chem.scand.38b-0049.
- ^ Lockner, James. "Stoichiometric Enamine Chemistry" (PDF). Baran Group, The Scripps Research Institute. Retrieved 26 November 2014.
- ^ White, William Andrew; Weingarten, Harold (January 1967). "A versatile new enamine synthesis". The Journal of Organic Chemistry. 32 (1): 213–214. doi:10.1021/jo01277a052.
- ^ Wade, L.G. (1999). Organic Chemistry. Saddle River, NJ: Prentice Hall. pp. 1019. ISBN 9780139227417.
- ^ Farmer, Steven (2013-10-16). "Enamine Reactions". UC Davis Chem Wiki.
- ^ Evans, D. "Enolates and Metalloenamines II" (PDF). Retrieved 10 December 2014.[영구적 데드링크]
- ^ Meyers, A. I.; Williams, Donald R. (August 1978). "Asymmetric alkylation of acyclic ketones via chiral metallo enamines. Effect of kinetic vs. thermodynamic metalations". The Journal of Organic Chemistry. 43 (16): 3245–3247. doi:10.1021/jo00410a034.
- ^ Seufert, Walter; Eiffenberger, Franz (1979). "Zur Halogenierung von Enaminen — Darstellung von β-Halogen-iminium-halogeniden". Chemische Berichte. 112 (5): 1670–1676. doi:10.1002/cber.19791120517.
- ^ Ito, Y; Konoike, T; Saegusa, T (1975). "Synthesis of 1,4-diketones by the reaction of silyl enol ether with silver oxide. Regiospecific formation of silver(I) enolate intermediates". Journal of the American Chemical Society. 97 (3): 649–651. doi:10.1021/ja00836a034.
- ^ Jang, HY; Hong, JB; MacMillan, DWC (2007). "Enantioselective organocatalytic singly occupied molecular orbital activation: the enantioselective alpha-enolation of aldehydes" (PDF). J. Am. Chem. Soc. 129 (22): 7004–7005. doi:10.1021/ja0719428. PMID 17497866.
- ^ Li, Q; Fan, A; Lu, Z; Cui, Y; Lin, W; Jia, Y (2010). "One-pot AgOAc-mediated synthesis of polysubstituted pyrroles from primary amines and aldehydes: application to the total synthesis of purpurone". Organic Letters. 12 (18): 4066–4069. doi:10.1021/ol101644g. PMID 20734981.
- ^ Guo, Fenghai; Clift, Michael D.; Thomson, Regan J. (September 2012). "Oxidative Coupling of Enolates, Enol Silanes, and Enamines: Methods and Natural Product Synthesis". European Journal of Organic Chemistry. 2012 (26): 4881–4896. doi:10.1002/ejoc.201200665. PMC 3586739. PMID 23471479.
- ^ List, Benjamin (2002). "Proline-catalyzed asymmetric reactions". Tetrahedron. 58 (28): 5573–5590. doi:10.1016/s0040-4020(02)00516-1.
- ^ Bui, Tommy; Barbas (2000). "A proline-catalyzed asymmetric Robinson Annulation". Tetrahedron Letters. 41 (36): 6951–6954. doi:10.1016/s0040-4039(00)01180-1.
- ^ Wiener, Jake. "Enantioselective Organic Catalysis:Non-MacMillan Approaches" (PDF). Archived from the original (PDF) on 26 October 2017. Retrieved 29 November 2014.
- ^ Hickmott, Peter (May 1982). "Enamines: Recent advances in synthetic, spectroscopic, mechanistic, and stereochemical aspects—II". Tetrahedron. 38 (23): 3363–3446. doi:10.1016/0040-4020(82)85027-8.
- ^ Mayr, H. (2003). "Structure-Nucleophilicity Relationships for Enamines". Chem. Eur. J. 9 (10): 2209–18. doi:10.1002/chem.200204666. PMID 12772295.
- ^ Hickmott, Peter (May 1982). "Enamines: Recent advances in synthetic, spectroscopic, mechanistic, and stereochemical aspects—II". Tetrahedron. 38 (23): 3363–3446. doi:10.1016/0040-4020(82)85027-8.
- ^ Lockner, James. "Stoichiometric Enamine Chemistry" (PDF). Baran Group, The Scripps Research Institute. Retrieved 26 November 2014.